Agencia Estatal Boletín Oficial del Estado






 Este texto consolidado es de carácter informativo y no tiene valor jurídico.
Este texto consolidado es de carácter informativo y no tiene valor jurídico.Incluye corrección de errores publicada en BOE núm. 219, de 13 de septiembre de 1989. Ref. BOE-A-1989-22117.
[Bloque 2: #preambulo]
El Decreto de Presidencia del Gobierno 2484/1967, de 21 de septiembre («Boletín Oficial del Estado» de 17 al 23 de octubre), por el que se aprueba el texto del Código Alimentario Español, prevé que puedan ser objeto de Reglamentaciones especiales las materias en él reguladas.
Por Decreto 2181/1975, de 12 de septiembre («Boletín Oficial del Estado» del 13), se aprobó la Reglamentación Técnico-Sanitaria para la elaboración, circulación y comercio de Pastas Alimenticias y, juntamente con ésta, se aprobaron los métodos oficiales de análisis para las mismas.
En el período de tiempo transcurrido, los avances llevados a cabo en el campo de la tecnología han sido lo suficientemente importantes para permitir una mayor precisión en la medición de componentes de los diferentes productos, posibilitando así el que puedan llegar a fijarse límites tolerables más estrictos, si se estimara necesario.
Este hecho, así como algunas innovaciones en la metodología analítica, hacen ahora aconsejable modificar los métodos oficiales aprobados por el citado Decreto.
En la redacción de los métodos oficiales de análisis se ha procurado, en lo posible, su adaptación a los métodos aprobados por los Organismos internacionales especializados en la materia, con el fin de aprovechar la experiencia obtenida de su aplicación, dado que, por otra parte, la Comunidad Económica Europea no tiene legislación específica aplicable en este sentido.
En su virtud, a propuesta de los Ministros de Economía y Hacienda, de Industria y Energía, de Agricultura, Pesca y Alimentación y de Sanidad y Consumo, oídos los sectores afectados, de conformidad con el informe preceptivo de la Comisión Interministerial para la Ordenación Alimentaria, y previa deliberación del Consejo de Ministros en su reunión del día 19 de junio de 1987,
DISPONGO:
[Bloque 3: #a1]
Se aprueban como oficiales los métodos de análisis para pastas alimenticias que se citan en el anejo I.
[Bloque 4: #a2-2]
Cuando existan métodos oficiales para determinados análisis, y hasta tanto los mismos no sean propuestos por el órgano competente y previamente informados por la Comisión Interministerial para la ordenación alimentaria, podrán ser utilizados los aprobados por los Organismos nacionales o internacionales de reconocida solvencia.
[Bloque 5: #dd]
Quedan derogados a la entrada en vigor de este Real Decreto el anejo del Decreto 2181/1975, de 12 de septiembre, por el que se aprueba la Reglamentación Técnico-Sanitaria para la elaboración, circulación y comercio de Pastas Alimenticias; el artículo 5.° del Real Decreto 1771/1976, de 2 de julio («Boletín Oficial del Estado» del 28), por el que se modifican algunos de los artículos y epígrafes de determinadas Reglamentaciones Técnico-Sanitarias y normas alimentarias específicas, así como cuantas disposiciones de igual o inferior rango se opongan a lo dispuesto en el mismo.
[Bloque 6: #dfprimera]
Los Ministerios proponentes quedan autorizados para que, a propuesta de los Organismos competentes, mediante Orden, y previo informe preceptivo de la Comisión Interministerial para la Ordenación Alimentaria, puedan dictar las disposiciones precisas para modificar, en caso necesario, los métodos oficiales de análisis para pastas alimenticias que se citan en el anejo I.
[Bloque 7: #dfsegunda]
El presente Real Decreto entrará en vigor a los treinta días de su publicación en el «Boletín Oficial del Estado».
[Bloque 8: #firma]
Dado en Madrid a 19 de junio de 1987.
JUAN CARLOS R.
El Ministro de Relaciones con las Cortes y de la Secretaría del Gobierno,
VIRGILIO ZAPATERO GÓMEZ
[Bloque 9: #anejoi]
1. Preparación de la muestra.
2. Humedad.
3. Cenizas.
4. Grasa.
5. Proteínas.
6. Fibra alimentaria insoluble.
7. Fibra bruta.
8. Azúcares.
9. Cloruros.
10. Acidez de la grasa.
11. Plomo.
12. Mercurio.
13. Cobre.
14. Arsénico.
1. Preparación de la muestra
1.1 Principio.
Homogeneización y reducción de la muestra al tamaño adecuado para la correcta realización del análisis.
1.2 Material y aparatos.
1.2.1 Aparato triturador que no provoque calentamiento, fácil de limpiar, y que proporcione un tamaño de partículas comprendido entre 800 y 1.200 µ.
1.2.2 Envases de capacidad suficiente, con cierre hermético, para conservar la muestra.
1.3 Procedimiento.
1.3.1 Muestra contenida en un solo envase.
Homogeneizar la muestra. Tomar un mínimo de 200 g y triturarlo en el aparato descrito en 1.2.1 y volver a homogeneizar.
1.3.2 Muestra contenida en varios envases.
Homogeneizar la porción de muestra contenida en cada envase, tornar de cada uno cantidades iguales para obtener finalmente un mínimo de 200 g de muestra. Triturar en el aparato descrito en 1.2.1 y volver a homogeneizar.
1.4 Observaciones.
Preparada la muestra, ésta servirá de base a todas las determinaciones, salvo mención expresa en contra, procurando realizar la preparación de los análisis en el menor tiempo posible.
2. Humedad
2.1 Principio.
Se determina la pérdida de peso de la muestra al someterla a calentamiento en estufa en condiciones determinadas.
2.2 Material y aparatos.
2.2.1 Balanza analítica con precisión de 0,1 mg.
2.2.2 Pesasustancias metálico o de vidrio con tapadera y con una superficie útil que permita un reparto de la muestra de 0,3 g/cm2 como máximo.
2.2.3 Estufa isoterma de calefacción eléctrica, a ser posible de aire forzado, regulada de tal manera que la temperatura del aire en su interior sea de 130° C y que tenga aireación suficiente. La estufa tendrá una capacidad calorífica tal que, regulada previamente a la temperatura de 130° C, pueda alcanzar de nuevo esa temperatura en menos de media hora, después de colocar simultáneamente en su interior el número máximo de muestras a desecar.
La eficacia de la ventilación se determinará con la ayuda de sémola como material de ensayo, que tenga un milímetro como máximo de partícula. La ventilación será tal que, secando simultáneamente a 130° C todas las muestras que la estufa pueda contener, primero durante dos horas y después durante tres horas, los resultados presenten entre ellos una diferencia inferior a 0,15 por 100 en valor absoluto.
2.2.4 Desecador provisto de un deshidratante eficaz.
2.3 Procedimiento.
Pesar con precisión de 1 mg, aproximadamente 5 g de muestra, en pesasustancias previamente preparada según método número 1.
Introducir el pesasustancias en la estufa (2.2.3) a 130 ± l° C y destapar. Mantener en la estufa durante una hora y cinco minutos. Tapar el pesasustancias antes de sacar de la estufa y dejar enfriar a temperatura ambiente en desecador y pesar a continuación.
2.4 Cálculos.
La humedad de la muestra expresada en tanto por ciento vendrá dada por la siguiente fórmula:
|
H%= |
(P1 – P2) 100 |
|
P |
Siendo:
P1 = Peso, en g, del pesasustancias con la muestra.
P2 = Peso, en g, del pesasustancias con la muestra desecada.
P = Peso, en g, de la muestra.
La diferencia resultante entre determinaciones duplicadas de la misma muestra no deberá ser mayor de 0,1 por 100 en valor absoluto.
2.5 Referencias.
1. Métodos de la Asociación Internacional de Química Cerealista (ICC).
2. ADAC M. 14.003 1980.
3. Cenizas
3.1 Principio.
Residuo obtenido por incineración a una temperatura de 550 ± 10° C hasta combustión completa de la materia orgánica y obtención de un peso constante.
3.2 Material y aparatos.
3.2.1 Crisoles no atacables en las condiciones del ensayo, con unas dimensiones mínimas de 40 mm de altura y 45 mm de diámetro superior.
3.2.2 Placa calefactora.
3.2.3 Horno eléctrico (mufla) con dispositivo de control de temperatura.
3.2.4 Desecador capaz de contener un deshidratante eficaz.
3.2.5 Varillas de vidrio con una extremidad aplanada.
3.2.6 Balanza analítica, con precisión de 0,1 mg.
3.3 Procedimiento.
Pesar con la precisión de un mg de 2 a 6 g de muestra preparada según el método oficial número 1, en un crisol previamente incinerado y tarado.
Colocar el crisol y su contenido sobre una placa calefactora, teniendo cuidado de que la combustión no sea demasiado rápida, de manera que no haya pérdidas de materia sólida por proyección. Llevar a continuación el crisol a la mufla (550 ± 10° C) hasta combustión completa de la sustancia (cenizas blancas o grises).
Enfriar a temperatura ambiente en un desecador.
Pesar seguidamente.
3.4 Cálculos.
3.4.1 El contenido en cenizas sobre sustancia natural vendrá dado por la siguiente fórmula:
|
% cenizas = |
P1 – P2 |
• 100 |
|
P |
Siendo:
P1 = Peso, en gramos, del crisol con las cenizas.
P2 = Peso, en gramos, del crisol vacío.
P = Peso, en gramos, de la muestra.
3.4.2 El contenido en cenizas sobre sustancia seca vendrá dado por la siguiente fórmula:
|
% cenizas = |
C × 100 |
|
100 – H |
Siendo:
C = % de cenizas obtenidas en (3.4.1).
H = Humedad.
En ambos casos los resultados se darán considerando solamente la primera cifra decimal.
3.5 Observaciones.
3.5.1 En caso necesario, para obtener una incineración uniforme puede humedecerse la muestra antes de la preincineración con etanol del 95 por 100 o aceite vegetal exento de cenizas.
3.5.2 Si la muestra a analizar contiene cloruros añadidos, deducir del valor de cenizas obtenido por el procedimiento anterior el porcentaje correspondiente de los mismos.
3.5.3 Límite de errores. Cuando el contenido de cenizas no rebase el 1 por 100 de la muestra, la diferencia de los resultados de un ensayo efectuado por duplicado no deberá ser superior al 0,02 por 100. Si el contenido de cenizas rebasa el 1 por 100 la diferencia no deberá ser superior al 2 por 100 de dicho contenido. Si es superior se repetirá la determinación.
3.6 Referencias.
1. ADAD, edición 1980, 14.006.
4. Grasa
4.1 Principio.
El producto es hidrolizado con ácido clorhídrico diluido. La masa seca conteniendo las materias grasas son extraídos con éter, el solvente evaporado y el residuo pesado.
4.2 Reactivos.
4.2.1 Ácido clorhídrico 3 N,
4.2.2 Éter etílico exento de peróxidos.
4.2.3 Solución de nitrato de plata.
4.3 Material y aparatos.
4.3.1 Extractor tipo Soxhlet.
4.3.2 Estufa de desecación capaz de mantener constante la temperatura a 100° C ± 1° C.
4.3.3 Desecador provisto de un deshidratante eficaz.
4.4 Procedimiento.
Pesar, con la precisión de un mg, aproximadamente 10 g de muestra preparada según el método oficial número 1 en un matraz de 250 a 300 ml.
Agitando continuamente añadir 100 ml de ácido clorhídrico (4.2.1), añadir unas perlas de vidrio o piedra pómez lavada y seca y cerrar con tapón de vidrio que no ajuste herméticamente o vidrio de reloj. Hervir unos sesenta minutos, agitando de cuando en cuando, enfriar y filtrar sobre filtro previamente humedecido. Lavar el precipitado con agua destilada hasta que el filtrado no dé precipitado con nitrato de plata o no dé reacción ácida de papel de tornasol.
Poner el filtro en una cápsula secar en estufa a 100° 0 ± 1°C.
El filtro ya seco se introduce en un cartucho para extractor tipo Soxhlet y se tapa con algodón desengrasado. El cartucho se coloca en el extractor y se vierte el éter etílico, dejándolo sifonar unas ocho horas.
El matraz receptor debe estar secado y tarado.
Evaporar el solvente, secar en estufa y pesar.
4.5 Cálculos.
4.5.1 El contenido de grasa en sustancia natural vendrá dado por la siguiente fórmula:
|
% grasas = |
P1 – P2 |
• 100 |
|
P |
Siendo:
P1 = Peso, en gramos, del matraz con la grasa.
P2 = Peso, en gramos, del matraz vacío.
P = Peso, en gramos, de la muestra.
4.5.2 El contenido de grasas en sustancia seca vendrá dado por la siguiente fórmula:
|
% grasa = |
G × 100 |
|
100 – H |
Siendo:
G = Porcentaje de grasa obtenida en 4.5.1.
H = Humedad.
4.6 Observaciones.
4.6.1 Para efectuar el procedimiento anterior podrán utilizarse sistemas automáticos o semiautomáticos, adaptándose a las especificaciones del equipo.
4.7 Referencias.
1. ADAC, edición 1980, 14.059.
5. Proteínas
5.1 Principio.
Determinación del nitrógeno, convirtiendo el nitrógeno orgánico presente en sulfato amónico con ácido sulfúrico. Después de alcalinizar con hidróxido sódico, destilar recogiendo el destilado sobre ácido bórico, titulando el amoniaco recogido con ácido N/10.
5.2 Reactivos.
5.2.1 Ácido sulfúrico 93, 99 por 100 libre de nitrógeno.
5.2.2 Hidróxido sódico al 40 por 100.
5.2.3 Catalizador. (Mezclar 5 de sulfato sódico o potásico con 5 mg de selenio.) También puede utilizarse otro catalizador adecuado.
5.2.4 Indicador de fenolftaleína al 1 por 100 en alcohol etílico.
5.2.5 Indicador Taschiro. Mezclar 20 mg de rojo de metilo y 10 mg de azul de metileno en 100 ml de alcohol etílico. También puede utilizarse rojo de metilo preparado en la de proporción 0,5 por 100 en alcohol etílico.
5.2.6 Solución de ácido bórico al 4 por 100.
5.2.7 Solución de ácido sulfúrico o clorhídrico N/10.
5.3 Material y aparatos.
5.3.1 Para digestión.
5.3.1.1 Matraces tipo Kjeldahl o similar.
5.3.1.2 Batería de mantas eléctricas o similar.
5.3.2 Para destilación.
5.3.2.1 Matraz generador de vapor.
5.3.2.2 Refrigerante.
5.3.2.3 Matraz receptor.
5.3.3 Titulación.
5.3.3.1 Bureta de vidrio o bureta automática.
5.4 Procedimiento.
Pesar, con la precisión de 1 mg, aproximadamente 0,5-2,5 g de muestra, preparada según el método oficial número 1, introducirla en el matraz Kjeldahl (5.3.1.1). Añadir unos 5 g del catalizador (5.2.3), 20 ml de ácido sulfúrico (5.2.1) (la cantidad varía según contenido en proteínas y grasa de la muestra). Poner a digerir en 5.3.1.2, teniendo cuidado al principio de no elevar demasiado la temperatura hasta que cese el desprendimiento de la espuma (añadir si fuera preciso una pequeña cantidad de parafina). Digerir hasta que la solución esté clara. Enfriar, diluir, añadir unas gotas de fenolftaleína (5.2.4) y conectar al aparato destilador añadiendo hidróxido sódico (5.2.2) hasta virage.
En el matraz receptor poner 100 ml de ácido bórico (5.2.6) con unas gotas de indicador (5.2.5), cuidando que el extremo del refrigerante quede bien cubierto del líquido.
Mantener la destilación aproximadamente 15 minutos (o más, si es preciso), hasta que no dé reacción básica); lavar el extremo del refrigerante y titular el destilado con ácido sulfúrico o clorhídrico N/10 (5.2.7).
Hacer un blanco.
5.5 Cálculos.
5.5.1 El contenido de proteínas en materia natural vendrá dado por la siguiente fórmula:
|
% proteínas = |
0,14 × 6,25 (V1 - V0) |
|
P |
Siendo:
V1 = Volumen, en mililitros, de ácido clorhídrico o sulfúrico utilizado en la determinación.
V0 = Volumen, en mililitros, de ácido clorhídrico o sulfúrico utilizado en blanco.
P = Peso, en gramos, de la muestra.
5.5.2 El contenido en proteínas en materia seca vendrá dado por la siguiente fórmula:
|
% proteínas = |
p × 100 |
|
100 – H |
Siendo:
p = % proteína obtenida en 5.5.1.
H = Humedad.
5.6 Observaciones.
5.6.1 La diferencia entre dos determinaciones sucesivas expresada en % de proteínas no debe ser superior al 0,25 por 100.
5.6.2 Para efectuar el procedimiento Kjeldahl podrán utilizarse sistemas automáticos o semiautomáticos, adaptándose a las especificaciones del equipo.
5.7 Referencias.
1. ADAC (1980) 2.057.
2. Pearson, 5.ª edición (1962).
6. Fibra alimentaria insoluble
6.1 Principio.
La muestra se extrae con una solución de detergente neutro en caliente. El residuo se incuba con una solución amilásica y se filtra. La determinación de las cenizas en el residuo filtrado permite conocer, por diferencia de peso, la cantidad de celulosa, hemicelulosa y lignina de la muestra.
6.2 Material.
6.2.1 Baño termostatizado y refrigerante de reflujo.
6.2.2 Filtros de vidrio fritado del número 2.
6.2.3 Sistema de filtración por succión a vacío.
6.2.4 Desecador.
6.2.5 Estufa para 37 grados y 110 grados centígrados.
6.2.6 Horno eléctrico (mufla) con dispositivo de control de ten peratura.
6.2.7 Balanza de precisión.
6.3 Reactivos.
6.3.1 Lauril sulfato sódico.
6.3.2 EDTA disódico dihidratado.
6.3.3 Tetraborato de sodio decahidratado (B407Na2.10 H20).
6.3.4 2-Etoxietanol.
6.3.5 Decahidronaftaleno.
6.3.6 Sulfito de sodio.
6.3.7 Fosfato disódico anhidro (PO4HNa2).
6.3.8 Fosfato monosódico anhidro (PO4H2Na).
6.3.9 Acetona.
6.3.10 α-Amilasa tipo VI-A (sigma A-6880 o equivalente).
6.3.11 Ácido fosfórico.
6.3.12 Solución de detergente neutro: Mezclar 18,61 gramos de EDTA disódico y 6,81 gramos de tetraborato de sodio decahidrato con 150 mililitros de agua y calentar hasta su disolución. Disolver 30 gramos de lauril sulfato sódico y 10 mililitros de 2-etoxietanol en 700 mililitros de agua caliente y mezclar con la solución anterior. Disolver 4,56 gramos de fosfato disódico anhidro en 150 mililitros de agua y mezclar con las soluciones anteriores. Ajustar a pH 6,9 - 7 con ácido fosfórico, si fuera necesario.
6.3.13 Solución tampón fosfato 0,1 N: Mezclar 39,2 mililitros de fosfato monosódico anhidro 0,1 M (preparado disolviendo 13,6 gramos en un litro de agua) con 60,8 mililitros de fosfato disódico 0,1 M (preparado disolviendo 14,2 gramos en un litro de agua).
6.4 Procedimiento.
Pesar, con precisión de 1 miligramo, aproximadamente 1 gramo de muestra preparada según método 1. Agregar ordenadamente 100 mililitros de solución de detergente neutro, 2 mililitros de decahidronaftaleno y 0,5 gramos de sulfito de sodio (6.3.6). Calentar hasta ebullición y mantener a reflujo durante una hora. Filtrar a través de filtro de vidrio fritado del número 2 (previamente calcinado a 550 grados centígrados) conectado a un sistema de succión por vacío.
Lavar sucesivamente unos 300 mililitros de agua hirviendo. Añadir hasta sobrepasar el nivel del residuo, una solución al 2,5 por 100 de -amilasa- en tampón fosfato 0,1 N.
Incubar a 37 grados centígrados durante dieciocho horas, aproximadamente.
Filtrar la solución enzimática por succión a través de un sistema de vacío y lavar el residuo con unos 80 mililitros de acetona. Secar el filtro con el residuo a 110 grados centígrados durante ocho horas, como mínimo. Enfriar en desecador y pesar.
Mantener el filtro con el residuo en mufla a 550 grados centígrados durante tres horas. Enfriar y pesar.
6.5 Cálculos.
El contenido en fibra alimentaria insoluble expresado en % vendrá dado por la siguiente fórmula:
|
% fibra alimentaria insoluble = |
P1 – P2 |
× 100 |
|
P0 |
Siendo:
P0 = Peso en miligramos de la muestra.
P1 = Peso en miligramos de crisol más residuo desecado a 110 grados centígrados.
P2 = Peso en miligramos del crisol más residuo calcinado.
6.6 Observaciones.
6.6.1 Las muestras conteniendo más de un 10 por 100 de materia grasa deberán desengrasarse previamente.
6.6.2 Para utilizar el procedimiento anterior podrán utilizarse sistemas automáticos o semiautomáticos adaptándose a las especificaciones del equipo.
6.7 Referencias.
1. Método AACC, 32-20 (1979).
7. Fibra bruta
7.1 Principio.
Tratar la muestra, desengrasada si es necesario, con soluciones de ácido sulfúrico e hidróxido potásico de concentraciones conocidas. Separar el residuo por filtración, lavar, desecar y pesar el residuo insoluble, determinando posteriormente su pérdida de masa por calcinación a 550 grados centígrados.
7.2 Material y aparatos.
7.2.1 Material de vidrio de uso corriente en el laboratorio.
7.2.2 Crisol filtrante número 2.
7.2.3 Horno de mufla con termostato.
7.2.4 Desecador provisto de un deshidratante eficaz.
7.2.5 Estufa capaz de mantener constante la temperatura de 130 grados centígrados ± 1 grado centígrado.
7.2.6 Equipo filtrante.
7.3 Reactivos.
7.3.1 Solución de sulfúrico 0,26 N. Disolver 1,25 gramos de ácido sulfúrico de d = 1,84 y riqueza 96 por 100 en 100 mililitros de agua.
7.3.2 Antiespumante.
7.3.3 Solución de hidróxido potásico 0,23 N: Disolver 1,25 gramos de hidróxido de potasio en 100 mililitros de agua.
7.3.4 Acetona pura.
7.3.5 Éter dictílico puro.
7.4 Procedimiento.
Pesar, con precisión de un miligramo, de 1 a 3 gramos de muestra y añadir 200 mililitros de ácido sulfúrico 0,26 N y unas gotas de antiespumante. Llevar a ebullición y mantenerla durante treinta minutos en un sistema de refrigeración a reflujo.
Transcurridos los treinta minutos filtrar sobre el crisol (7.2.2), previamente incinerado y lavar el residuo con agua caliente hasta que no dé reacción ácida.
Transferir cuantitativamente el residuo a un matraz adaptable al sistema de reflujo, añadir 200 mililitros de solución de hidróxido potásico 0,23 N y unas gotas de antiespumante. Llevar a ebullición y dejar hervir durante treinta minutos. Filtrar sobre crisol filtrante y lavar con agua caliente hasta que no dé reacción alcalina. Deshidratar lavando tres veces con acetona usando un volumen total de unos 100 mililitros.
Llevar el crisol a la estufa y secarlo a 130 grados centígrados durante dos horas.
Dejar enfriar en desecador y pesar rápido. Introducir a continuación el crisol en el horno (7.2.3) y dejar calcinar durante tres horas como mínimo a 550 grados centígrados. Dejar enfriar en desecador y pesar rápidamente.
7.5 Cálculos.
|
Fibra bruta (%) = 100 |
P1 – P2 |
|
P0 |
Siendo:
P0 = Peso inicial de la muestra.
P1 = Peso del crisol conteniendo la muestra desecada.
P2 = Peso del crisol conteniendo la muestra calcinada.
7.6 Observaciones.
7.6.1 Las muestras conteniendo más de un 10 por 100 de materia grasa deben desengrasarse con éter etílico antes del análisis.
7.6.2 Para efectuar el procedimiento anterior podrán utilizarse sistemas automáticos o semiautomáticos, adaptándose a las especificaciones del equipo.
7.7 Referencias.
1. Journal Official del Communautes Europeennes, número L 83/24, 1973.
8. Azúcares
8.1 Principio.
Eliminación de todas las materias reductoras distintas de los azúcares, mediante defecación a partir de las soluciones de Carrez I, II, previa disolución de los azúcares en etanol diluido. Eliminación del etanol y valoración antes y después de la inversión según el método de Luff-Schoorl.
8.2 Material y aparatos.
8.2.1 Agitador mecánico.
8.2.2 Matraces aforados de 1.000, 300, 200, 100 y 50 mili-litros.
8.3 Reactivos.
8.3.1 Etanol al 40 por 100 (v/v) d = 0,948 a 20° C.
8.3.2 Solución de Carrez I. Disolver en agua 24 gramos de acetato de cinc trihidrato y 3 mililitros de ácido acético glacial y añadir agua destilada hasta 100 mililitros.
8.3.3 Solución de Carrez II. Disolver en agua 10,6 de ferrocianuro potásico K4(FeCN6) . 3H2O y añadir agua destilada hasta 100 mililitros.
8.3.4 Solución de rojo de metilo al 0,1 por 100 (v/v).
8.3.5 Ácido clorhídrico 4 N.
8.3.6 Ácido clorhídrico 0,1 N.
8.3.7 Solución de hidróxido sódico 0,1 N.
8.3.8 Solución de sulfato de cobre. Disolver 25 gramos de sulfato de cobre (II) pentahidrato CuSO4 . 5H2O, exento de hierro, en agua y enrasar a 100 mililitros.
8.3.9 Solución de ácido cítrico. Disolver 50 gramos de ácido cítrico monohidrato C6H8O7 . H2O en 50 mililitros de agua.
8.3.10 Solución de carbonato de sodio. Disolver 143,8 gramos de carbonato de sodio anhidro en unos 300 mililitros de agua caliente, dejar enfriar y completar a 300 mililitros.
8.3.11 Solución de tiosulfato de sodio 0,1 N.
8.3.12 Solución de almidón. Añadir una mezcla de 5 gramos de almidón soluble en 30 mililitros de agua a 1 litro de agua hirviendo. Dejar hervir durante tres minutos. Dejar enfriar. Añadir 10 miligramos de ioduro de mercurio (II) como agente conservador.
8.3.13 Ácido sulfúrico a 6 N.
8.3.14 Solución de ioduro potásico al 30 por 100 (p/v).
8.3.15 Piedra pómez lavada con ácido clorhídrico y aclarada con agua.
8.3.16 Isopentanol.
8.3.17 Reactivo de Luff-Schoorl: Verter agitando cuidadosamente la solución de ácido cítrico (8.3.9) en la solución de carbonato de sodio (8.3.10). Agitar hasta la desaparición del desprendimiento gaseoso. A continuación añadir la solución de sulfato de cobre (II) (8.3.8) y completar hasta 1 litro con agua. Dejar reposar doce horas y filtrar. Verificar la normalidad del reactivo obtenido (Cu 0,1 N; Na2CO3 2N). El pH de la solución deber ser aproximadamente 9,4.
8.4 Procedimiento.
8.4.1 Preparación de la muestra. Pesar con aproximación de 1 miligramo, 2,5 gramos de la muestra e introducirlo en una matraz aforado de 250 mililitros. Añadir 200 mililitros de etanol al 40 por 100 (v/v) y mezclar durante una hora en el agitador. Añadir 5 mililitros de la solución Carrez I y agitar durante un minuto, adicionar y agitar durante el mismo tiempo con 5 mililitros de la solución Carrez II.
Enrasar a 250 mililitros con la solución de etanol (8.3.1), homogeneizar y filtrar. Tomar 200 mililitros de filtrado y evaporar aproximadamente hasta la mitad del volumen, a fin de eliminar la mayor parte del etanol. Trasvasar en su totalidad el residuo de evaporación, con ayuda de agua caliente, a un matraz aforado de 200 mililitros y enfriar, a continuación enrasar con agua y filtrar si es necesario. Esta solución será utilizada para la determinación de azúcares reductores y, después de la inversión, para la determinación de azúcares totales.
8.4.2 Determinación de azúcares reductores. Tomar como máximo 25 mililitros de la solución preparada según 8.4.1 y que contenga menos de 60 miligramos de azúcares reductores, expresado en glucosa. Si es necesario, completar el volumen hasta 25 mililitros con agua destilada y determinar la cantidad de azúcares reductores según Luff-Schoorl. El resultado será expresado en tantos por ciento de glucosa.
8.4.3 Determinación de azúcares totales previa inversión. Tomar 50 mililitros de la solución (8.4.1) y llevar a una matraz aforado de 100 mililitros. Añadir unas gotas de la solución rojo de metilo y adicionar lentamente agitando 15 mililitros de la solución de ácido clorhídrico 4 N hasta viraje a rojo. Añadir 15 mililitros de ácido clorhídrico 0,1 N y sumergirlo en un baño de agua caliente a ebullición durante treinta minutos. Refrigerar hasta 20° C y añadir a continuación 15 mililitros de la solución de hidróxido sódico 0,1 N (8.3.7). Enrasar a 100 mililitros con agua y homogeneizar.
Tomar una cantidad que no exceda de 25 mililitros y contenga menos de 60 miligramos de azúcares reductores, expresado en glucosa. Si es necesario, completar el volumen hasta 25 mililitros con agua destilada y determinar la cantidad de azúcares reductores según Luff-Schoorl. El resultado será expresado en tantos por ciento de glucosa. De expresarlo en sacarosa, se debe multiplicar por el factor 0,95.
8.4.4 Valoración de Luff-Schoorl. Tomar 25 mililitros del reactivo Luff-Schoorl (8.3.17) y llevarlo a un erlenmeyer de 300 mililitros, añadir 25 mililitros exactamente medidos de la solución defecada de azúcares, adicionar un poco de piedra pómez y calentar agitando. Adaptar en seguida un refrigerante de reflujo sobre el erlenmeyer, a partir de este momento hacer hervir la solución y mantener en ebullición durante diez minutos exactamente. Refrigerar inmediatamente al chorro de agua fría durante cinco minutos y proceder a su valoración.
Añadir 10 mililitros de la solución de ioduro potásico (8.3.14), inmediatamente después y con cuidado, 25 mililitros de ácido sulfúrico 6 N (8.3.13). Valorar a continuación mediante la solución de tiosulfato de sodio (8.3.9) hasta la aparición de color amarillo, añadir en ese momento la solución de almidón y terminar de valorar.
Efectuar la misma valoración sobre una mezcla que contenga 25 mililitros, exactamente medidos, del reactivo de Luff-Schoorl, 25 mililitros de agua, 10 mililitros de la solución de ioduro de potasio (8.3.14) y 25 mililitros de la solución de ácido sulfúrico 6 N (8.3.13) sin llevar a ebullición.
8.5 Cálculos.
Establecer por medio de la tabla I la cantidad de glucosa en miligramos correspondiente a la diferencia entre las dos valoraciones, según los mililitros de tiosulfato de sodio 0,1 N gastados en cada una de las valoraciones.
Expresar el resultado en tanto por ciento de azúcares en la muestra.
8.6 Observaciones.
8.6.1 Es recomendable añadir aproximadamente 1 mililitro de isopentanol (sin tener en cuenta el volumen) antes de la ebullición, con el reactivo Luff-Schoorl para evitar la formación de espuma.
8.6.2 La diferencia entre la cantidad de azúcares totales después de la inversión, expresada en glucosa, y la cantidad de azúcares reductores, expresada igualmente en glucosa, multiplicada por 0,95 da la cantidad en tanto por ciento de sacarosa.
8.6.3 Para calcular la cantidad de azúcares reductores, excluyendo la lactosa, se puede determinar de las siguientes formas:
8.6.3.1 Para un cálculo aproximado, multiplicar por 0,675 la cantidad de lactosa obtenida, por determinación separada y restar el resultado obtenido por la cantidad en azúcares reductores.
8.6.3.2 Para el cálculo preciso de azúcares reductores, excluyendo la lactosa, es necesario partir de la misma muestra 8.4.1 para las dos determinaciones finales. Uno de los análisis es efectuado a partir de la solución obtenida en 8.4.1 y el otro sobre una parte de la solución obtenida para la valoración de la lactosa según el método para la determinación de lactosa.
En los casos 8.6.3.1 y 8.6.3.2 la cantidad de azúcares presentes se determinan según el método de Luff-Schoorl, expresado en miligramos de glucosa.
La diferencia entre los dos valores se expresa en tanto por ciento de la muestra.
8.7 Referencias.
1. Journal Officiel des Communautes Europennes, número L 155/32, 1971.
TABLA 1
Para 25 ml de reactivo Luff-Schoorl
|
Na2 S2 O3 0,1W |
Glucosa, fructosa azúcares invertido C6H12O6 |
Lactosa C12 H22 O11 |
Maltosa C12 H22 O11 |
|||
|---|---|---|---|---|---|---|
|
ml |
mg |
Diferencia |
mg |
Diferencia |
mg |
Diferencia |
|
1 |
2,4 |
2,4 |
3,6 |
3,7 |
3,9 |
3,9 |
|
2 |
4,8 |
2,4 |
7,3 |
3,7 |
7,8 |
3,9 |
|
3 |
7,2 |
2,5 |
11,0 |
3,7 |
11,7 |
3,9 |
|
4 |
9,7 |
2,5 |
14,7 |
3,7 |
15,6 |
4,0 |
|
5 |
12,2 |
2,5 |
18,4 |
3,7 |
19,6 |
3,9 |
|
6 |
14,7 |
2,5 |
22,1 |
3,7 |
23,5 |
4,0 |
|
7 |
17,2 |
2,6 |
25,8 |
3,7 |
27,5 |
4,0 |
|
8 |
19,8 |
2,6 |
29,5 |
3,7 |
31,5 |
4,0 |
|
9 |
22,4 |
2,6 |
33,2 |
3,8 |
35,5 |
4,0 |
|
10 |
25,0 |
2,6 |
37,0 |
3,8 |
39,5 |
4,0 |
|
11 |
27,6 |
2,7 |
40,8 |
3,8 |
43,5 |
4,0 |
|
12 |
30,3 |
2,7 |
44,6 |
3,8 |
47,5 |
4,1 |
|
13 |
33,0 |
2,7 |
48,4 |
3,8 |
51,6 |
4,1 |
|
14 |
35,7 |
2,8 |
52,2 |
3,8 |
55,7 |
4,1 |
|
15 |
38,5 |
2,8 |
56,0 |
3,9 |
59,8 |
4,1 |
|
16 |
41,3 |
2,9 |
59,9 |
3,9 |
63,9 |
4,1 |
|
17 |
44,2 |
2,9 |
63,8 |
3,9 |
68,0 |
4,2 |
|
18 |
47,1 |
2,9 |
67,7 |
4,0 |
72,2 |
4,3 |
|
19 |
50,0 |
3,0 |
71,7 |
4,0 |
76,5 |
4,4 |
|
20 |
53,0 |
3,0 |
75,7 |
4,1 |
80,9 |
4,5 |
|
21 |
56,0 |
3,1 |
79,8 |
4,1 |
85,4 |
4,6 |
|
22 |
59,1 |
3,1 |
83,9 |
4,1 |
90,0 |
4,6 |
|
23 |
62,2 |
|
88,0 |
|
94,6 |
|
9. Cloruros
9.1 Principio.
Los cloruros se solubilizan en agua, defecándose la solución si contienen materias orgánicas, posterior acidificación de la misma con ácido nítrico y precipitación de los cloruros con nitrato de plata. El exceso de nitrato se valora con una solución de sulfocianuro de amonio.
9.2 Material y aparatos.
9.2.1 Agitador de 35 a 40 r.p.m.
9.3 Reactivos.
9.3.1 Solución de sulfocianuro de amonio 0,1 N.
9.3.2 Solución de nitrato de plata 0,1 N.
9.3.3 Solución saturada de sulfato amónico-férrico.
9.3.4 Ácido nítrico, d = 1,38.
9.3.5 Éter etílico.
9.3.6 Acetona.
9.3.7 Solución de Carrez I: Disolver en agua 24 g de acetato de cinc Zn (CH3 COO)2 2H2O y 3 g de ácido acético glacial. Completar hasta 1.000 ml con agua.
9.3.8 Solución de Carrez II: Disolver en agua 10,6 g de ferrocianuro de potasio K4 [Fe(CN)6] • 3H2O. Completar a 100 ml con agua.
9.3.9 Carbón activo, exento de cloruros.
9.4 Procedimiento.
Pesar, con precisión de 1 mg, aproximadamente, 5 g de muestra e introducirla con 1 g de carbón activo en un matraz aforado de 500 ml. Añadir 400 ml de agua a 20 °C aproximadamente 5 ml de la solución de Carrez I, agitar y añadir seguidamente 5 ml de la solución de Carrez II. Agitar durante treinta minutos, enrasar, homogeneizar y filtrar.
Tomar de 25 a 100 ml de filtrado (con contenido en cloro inferior a 150 mg) e introducirlo en un erlenmeyer, diluir si es necesario, hasta 50 ml con agua. Añadir 5 ml de ácido nítrico, 20 ml de solución saturada de sulfato amónico férrico y dos gotas de la solución de sulfocianuro amónico, añadidas mediante una bureta llena hasta el trazo cero. Añadir seguidamente mediante una bureta la solución de nitrato de plata hasta un exceso de 5 ml. Añadir 5 ml de éter etílico y agitar fuertemente para recoger el precipitado. Valorar el exceso de nitrato de plata mediante la solución de sulfocianuro amónico hasta que el viraje a rojo oscuro persista durante un minuto.
9.5 Cálculos.
La cantidad de cloro (p) expresada en cloruro de sodio presente en el volumen del filtrado separado para la valoración, viene dada por la fórmula:
p = 5,845 (V1 – V2) mg
Siendo:
V1= Volumen, en ml, de solución de nitrato de plata añadida.
V2 = Volumen, en ml, de solución de sulfocianuro amónico 0,1 utilizados en la valoración.
Efectuar un ensayo en blanco sin la muestra a analizar y si consumo solución de nitrato de plata, 0,1 N restar este valor el volumen (V1 – V2).
Expresar el resultado en porcentaje de la muestra.
9.6 Observaciones.
9.6.1 Para los productos ricos en materias grasas, desengrasar previamente mediante éter etílico.
9.7 Referencias.
1. Journal Officiel des Communautes Europeennes. Núm. L 155/23-1971.
10. Grado de acidez de las pastas alimenticias
1. Principio
La acidez del extracto alcohólico de las pastas alimenticias se determina por titulación y se expresa en ml/de NaOH 1 N.
2. Material y aparatos
2.1 Molino de laboratorio, que sin calentar la muestra sea capaz de obtener partículas inferiores a 550 micras.
2.2 Filtro de filtración rápida.
3. Reactivos
3.1 Alcohol etílico de 95°, exento de peróxidos.
3.2 Alcohol etílico de 50°. Mezclar 100 ml de alcohol etílico de 95° con 96 ml de agua destilada.
3.3 Solución alcohólica de fenolftaleina al 1 por 100.
4. Procedimiento
Moler la muestra a analizar de modo que pase completamente a través de un tamiz de malla de 500 micras.
Pesar, con precisión de un miligramo, 4 g de producto, transfiriéndolo a un matraz erlenmeyer de 500 ml con tapón esmerilado y añadir 100 ml de alcohol etílico de 50 grados (3.2) previamente neutralizados a la fenolftaleina (3.4) con NaOH 0,02 N (3.3). Agitar vigorosamente, dejar en contacto con la solución alcohólica durante tres horas, agitando periódicamente.
Transcurridas las tres horas, filtrar a través del filtro 2.2.
Tomar del filtrado 50 ml y titular con NaOH 0,02N (3.3), empleando como indicador tres gotas de solución alcohólica de fenolftaleina al 1 por 100 (3.4).
5. Expresión de los resultados
Se define el grado de acidez (G) como el número de ml de hidróxido sódico 1 N necesario para neutralizar la acidez de 100 g de producto seco.
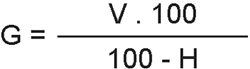
siendo: V = Volumen, en ml, de NaOH 0,02 N empleados en la neutralización.
H = Porcentaje de humedad de la muestra.
Expresar el resultado con una cifra decimal.
11. Plomo
11.1 Principio.
Determinación del plomo por A.A previa mineralización de la muestra.
11.1 Material y aparatos.
11.2.1 Espectrofotómetro de A.A.
11.2.2 Lámpara de plomo.
11.2.3 Cápsulas de platino, cuarzo o similar.
11.2.4 Baño de arena o placa calefactora.
11.2.5 Horno eléctrico (mufla) con dispositivo de control de temperatura.
11.3 Reactivos.
11.3.1 Ácido nítrico del 70 por 100 (d = 1,413).
11.3.2 Ácido nítrico al 1 por 100 en agua destilada (v/v).
11.3.3 Solución patrón de 1.000 mg de Pb/L. Disolver 1,598 g de (NO3)2 Pb enrasando a 1.000 mL con ácido nítrico al 1 por 100.
11.4 Procedimiento.
11.4.1 Preparación de la muestra. Poner 10 g de la muestra en la cápsula (11.2.3) llevar sobre la placa calefactora teniendo cuidado que la combustión no sea demasiado rápida de manera que no haya pérdidas de materia sólida por proyección. Añadir a continuación 2 ml de ácido nítrico y carbonizar el residuo en el baño de arena o plaza calefactora.
Seguidamente introducir la cápsula en la mufla y mantenerla a 450° C hasta mineralización total (11.6.1). Dejar enfriar. Disolver a continuación las cenizas con ácido nítrico concentrado y agua destilada. Llevar la solución a un matraz de 10 ml, lavar la cápsula con agua destilada y añadir las aguas de lavado hasta el enrase, filtrando posteriormente.
11.4.2 Construcción de la curva patrón.-Diluir alicuotas apropiadas de la solución patrón (11.3.3) con el ácido nítrico necesario para que su concentración sea similar a la dilución final de la muestra para obtener una curva de concentraciones 1, 2 y 3 mg/L.
11.4.3 Determinación.–Operar según las especificaciones del aparato; usando llama de aire-acetileno. Medir las abserbancias de la muestra y patrones a 283 nm. Si la solución está muy concentrada diluirla con ácido nítrico al 1 por 100.
11.5 Cálculos.
Calcular el contenido en plomo, expresado en mg/L mediante comparación con la correspondiente curva patrón y teniendo en cuenta el factor de dilución.
11.6 Observaciones.
11.6.1 En caso de que la muestra no esté totalmente mineralizada añadir unas gotas de ácido nítrico concentrado y repetir el proceso.
11.7 Referencias.
1. Métodos Oficiales de Análisis de Vinos. Ministerio de Agricultura, página 134 (I), 1976.
12. Mercurio
12.1 Principio.
Determinación de mercurio por absorción atómica con técnica de vapor frío previa digestión de la muestra.
12.2 Material y aparatos.
12.2.1 Balanza de precisión.
12.2.2 Espectrofotómetro de absorción atómica.
12.2.3 Lámpara de mercurio.
12.2.4 Cámara de absorción con ventanas de cuarzo acoplable al espectrofotómetro.
12.2.5 Equipo de reducción del ión mercurio a mercurio metálico y de arrastre hasta la cámara de absorción incluyendo sistema de desecación.
12.2.6 Registrador gráfico de voltaje y velocidad de carta variable.
12.2.7 Material de vidrio corriente de laboratorio lavado con ácido nítrico (1:1) y enjuagado con agua destilada.
12.2.8 Bloque de digestión con temperatura programable.
12.2.9 Tubos de digestión para el bloque anterior.
12.2.10 Tubos de condensación adaptables a los tubos de digestión.
12.3 Reactivos.
12.3.1 Ácido sulfúrico 95 - 97 por 100 (d = 1,84).
12.3.2 Agua oxigenada 18 por 100 p/V.
12.3.3 Ácido nítrico 70 por 100 (d = 1,41).
12.3.4 Cloruro estannoso (exento de mercurio) 10 por 100.
12.3.5 Agua destilada.
12.3.6 Solución patrón de mercurio concentrada de 1.000 mg/l. Disolver 0,1354 de cloruro mercúrico en 75 ml de agua desionizada, añadir 10 ml de ácido nítrico y ajustar el volumen a 100 ml.
12.3.7 Solución patrón de mercurio de 0,1 mg/1 se obtiene de la anterior por sucesivas diluciones con agua desionizada. Debe prepararse al igual que las soluciones intermedias diariamente.
12.4 Procedimiento.
12.4.1 Preparación de la muestra: Colocar de 3 a 5 g de muestra en un tubo digestor (12.29) acoplando a éste un tubo de condensación (12.2.10). Añadir 10 ml de ácido sulfúrico a incrementos de 1 ml (la muestra se carboniza; pero, si el ácido se añade lentamente, de modo que la temperatura de la solución permanezca baja y, se toma la precaución de que no se formen masas de carbón, el mercurio queda en solución). Añadir 10 ml de agua oxigenada (12.3.2) a incrementos de 1 ml. Dejar reaccionar antes de añadir el siguiente incremento. Añadir 10 ml de ácido nítrico e incrementos de 1 ml. Lavar los condensadores con agua destilada y retirarlos.
12.4.2 Digestión de la muestra: Colocar los tubos de digestión en el bloque calefactor y calentar a 100° C. Mantener esta temperatura durante 6 minutos, aumentarlo entonces hasta 200° C a razón de 4° C/minuto. Retirar los tubos del bloque y dejar enfriar. Transferir las soluciones a matraces de 100 ml y enrasar con agua destilada.
12.4.3 Determinación: Se analiza la muestra por la técnica de vapor frío A.A. según las instrucciones propias de cada aparato, utilizando cloruro estannoso como agente reductor (12.3.4) siendo la longitud de onda de medida 254 nm.
12.4.4 Construcción de la curva patrón: Se obtiene representando en abcisas los contenidos en mercurio de los patrones preparados con alicuotas de 0; 0,5; 1,0; 2,0; 5,0 y 10 ml de solución patrón de 0,1 mg/1 de forma que contengan de o a 1 mg de mercurio y en ordenadas a la altura de los correspondientes máximos de absorción. Dichos patrones se habrán sometido al mismo procedimiento que las muestras.
12.5 Cálculos.
Calcular el contenido en mercurio refiriéndolos a la curva patrón obtenida previamente y operando en idénticas condiciones. L
|
mg de Hg/Kg |
L |
|
P |
Siendo:
L = La lectura obtenida a partir de la gráfica expresada en microgramos.
P = Peso, en gramos, de la muestra.
12.6 Referencias.
1. Munns y Holland. JAOAC 60 833-837, 1977.
2. Marts y Blahc. JAOAC vol 66, número 6 1983.
3. AOAC Métodos oficiales de análisis, 1980.
13. Cobre
13.1 Principio.
Determinación del cobre por A.A. previa mineralización de la muestra.
13.2 Material y aparatos.
13.2.1 Esoectrofotómetro de A.A.
13.2.2 Lámpara de cobre.
13.2.3 Las utilizadas para el plomo, en (11.2.3), (11.2.4) y (11.2.5).
13.3 Reactivos.
13.3.1 Los utilizados para el plomo, en (11.3.1) y (11.3.2).
13.3.2 Solución patrón de 1.000 mg de Cu/L. Disolver 1.000 g de Cu puro en el mínimo volumen necesario de NO3H (1:1) y diluir a 1 litro con ácido nítrico del 1 por 100 (v/v).
13.4 Procedimiento.
13.4.1 Preparación de la muestra.
Como en (11.4.1).
13.4.2 Construcción de la curva patrón.–Diluir partes alícuotas de la solución patrón (13.3.2) con ácido nítrico del 1 por 100 para obtener soluciones que contengan a 1 a 5 mg de Cu/L.
13.4.3 Determinación.-Igual que para el plomo. Medir a 324,7 nm.
13.5 Cálculos.
Partiendo de los valores de absorbancia obtenidos para la muestra, hallar mediante la curva patrón las concentraciones de cobre de la muestra.
13.6 Referencias.
1. H. E. Parker: «Atomic Absorption Newsletter (1963), 13”».
2. F. Rousselet: «Spectrophotometrie para absorption atomique Boudin» Ed. París (1968), págs. 59-144.
14. Arsénico
14.1 Principio.
La muestra se somete a una digestión ácida con una mezcla de ácido nítrico y sulfúrico.
La determinación del arsénico se realiza por espectrofotometría de absorción atómica, con generador de hidruros.
14.2 Material y aparatos.
14.2.1 Balanza analítica con precisión de 0,1 mg.
14.2.2 Matraces Kjeldahl de 250 ml.
14.2.3 Espectrofotómetro de absorción atómica equipado con sistema generador de hidruros.
14.2.4 Lámpara de descarga sin electrodos.
14.2.5 Fuente de alimentación para lámpara de descarga sin electrodos.
14.2.6 Registro gráfico.
14.3 Reactivos.
Se utilizan solamente reactivos de grado de pureza para análisis y agua destilada.
14.3.1 Ácido clorhídrico (d = 1,19 g/ml).
14.3.2 Disolución de ácido clorhídrico 32 por 100 V/V.
Disolver 32 ml de ácido clorhídrico (d = 1,19 g/ml) con agua destilada hasta un volumen de 100 ml.
14.3.3 Disolución de ácido clorhídrico 1,5 por 100 V/V.
Disolver 15 ml de ácido clorhídrico (d =1,19 g/ml) con agua destilada hasta un volumen de 1.000 ml.
14.3.4 Ácido nítrico (d = 1,40).
14.3.5 Ácido sulfúrico (d = 1,84).
14.3.6 Disolución de hodróxido de sodio al 1 por 100.
Pesar 1 g de hidróxido de sodio y disolverlo con agua destilada hasta un volumen de 100 ml.
14.3.7 Disolución de borohidruro de sodio al 3 por 100.
Pesar 3 g de borohidruro de sodio y disolverlo hasta 100 ml con idróxido de sodio al 1 por 100.
14.3.8 Disolución de tritiplex III al 1 por 100.
Pesar 1 g de tritiplex III y disolverlo hasta 100 ml con agua destilada.
14.3.9 Disolución de hidróxido de potasio al 20 por 100.
Pesar 20 g de hidróxido de potasio y disolverlo con agua destilada hasta un volumen de 100 ml.
14.3.10 Disolución de ácido sulfúrico al 20 por 100 (V/V).
Diluir 20 ml de ácido sulfúrico (14.3.5) con agua destilada hasta un volumen de 100 ml.
14.3.11 Disolución de ácido sulfúrico al 1 por 100 (V/V).
Diluir 1 ml de ácido sulfúrico (14.3.5) con agua destilada hasta un volumen de 100 ml.
14.3.12. Solución patrón de arsénico de concentración 1 g/l.
Disolver 0,132 g de trióxido de arsénico en 2,5 ml de hidróxido de potasio al 20 por 100 (14.3.9), neutralizar con ácido sulfúrico al 20 por 100 (14.3.10), diluir hasta 100 ml con ácido sulfúrico 1 por 100 (14.3.11).
14.3.13. Solución patrón de arsénico de concentración 10 mg/l.
Pipetear 1 ml de la solución patrón de arsénico (14.3.12) en un matraz aforado de 100 ml. Diluir hasta el enrase con agua destilada.
14.3.14. Solución patrón de arsénico de concentración 0,1 mg/l.
Pipetear 1 ml de la solución de arsénico (14.3.13) en un matraz aforado de 100 ml. Diluir hasta el enrase con agua destilada.
14.4. Procedimiento.
14.4.1. Preparación de la muestra.
En un matraz Kjeldahl, de 250 ml, introducir 2 g de muestra con 20 ml de ácido nítrico (14.3.4) y 5 ml de ácido sulfúrico (14.3.5).
Llevar a ebullición hasta un volumen aproximado a 5 ml. Dejar enfriar y disolver con agua destilada en un matraz de 50 ml de la solución resultante.
14.4.2. Preparación del blanco y patrones de trabajo.
En un matraz Kjeldahl, introducir 5 ml de la solución de arsénico (14.3.14) y someterlo al mismo tratamiento que la muestra.
Un ml de la solución contiene 10 nanogramos de arsénico.
Preparar un blanco con todos los reactivos utilizados siguiendo el tratamiento dado a la muestra.
14.4.3. Condiciones del espectrofotómetro.
Encender la fuente de alimentación de las lámparas de descarga sin electrodos con el tiempo suficiente para que se estabilice la energía de la lámpara.
Encender el espectrofotómetro, ajustar la longitud de onda a 193,7 nm, colocando la rejilla de acuerdo con las condiciones del aparato.
Encender el generador de hidruros, colocando la temperatura de la celda a 900° C, esperando hasta que se alcance dicha temperatura.
Se ajustan las condiciones del generador de hidruros según las especificaciones del aparato.
Ajustar el flujo de Argón de acuerdo con las características del aparato.
Encender el registrador.
14.4.4. Determinación.
Las determinaciones de la concentración de arsénico se realizan por el método de adición de patrones, por medio de medidas duplicadas en el espectrofotómetro en las condiciones especificadas en (14.4.3), añadiendo al matraz de reacción 3 ml de la solución (14.3.8) usando como reductor la solución (14.3.7); como patrones internos se usan 10, 20 y 50 ng de As.
Lavar los matraces antes y después de cada uso, con ácido clorhídrico 1,5 por 100 (14.3.3).
Al construir la gráfica de adición hay que descontar el valor de absorbancia del blanco obtenido en las mismas condiciones anteriores, pero añadiendo 3 ml de la solución blanco.
En estas condiciones el límite de detección de la técnica es de 5 ng.
Se modifica el punto 10 por el art. 2 del Real Decreto 1534/1991, de 18 de octubre. Ref. BOE-A-1991-26242.
Redactado conforme a la corrección de errores publicada en BOE núm. 219, de 13 de septiembre de 1989. Ref. BOE-A-1989-22117.
Este documento es de carácter informativo y no tiene valor jurídico.
Ayúdenos a mejorar: puede dirigir sus comentarios y sugerencias a nuestro Servicio de atención al ciudadano
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid





