Butlletí Oficial de l'Estat






Contingut no disponible en valencià
 Este texto consolidado es de carácter informativo y no tiene valor jurídico.
Este texto consolidado es de carácter informativo y no tiene valor jurídico.[Disposición derogada]
Norma derogada, con efectos de 2 de enero de 2021, por la disposición derogatoria única.2.b) del Real Decreto 957/2020, de 3 de noviembre. Ref. BOE-A-2020-14960
El Real Decreto 1344/2007, de 11 de octubre, por el que se regula la farmacovigilancia de medicamentos de uso humano, en su artículo 19 apartado 2 dispone que la Agencia Española de Medicamentos y Productos Sanitarios establecerá un Comité de Coordinación de Estudios Posautorización. Dicho Comité contará con la participación de los representantes de todas las comunidades autónomas y de la Agencia Española de Medicamentos y Productos Sanitarios.
Así mismo, dicho artículo prevé que las administraciones sanitarias establecerán de común acuerdo las condiciones en las que se llevarán a cabo los estudios posautorización de tipo observacional, con la finalidad de favorecer aquellos que puedan contribuir al conocimiento del medicamento o a mejorar la práctica clínica, así como que las directrices de los procedimientos comunes que cada comunidad autónoma ejercerá en su ámbito competencial, serán objeto de debate en el citado comité.
En cumplimiento de lo anteriormente expuesto, el Comité de Coordinación de Estudios Posautorización, fue constituido con fecha 24 de septiembre de 2008, procediendo en ejercicio de sus funciones a adoptar unas directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano.
Mediante esta orden se publican las «Directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano», que establecen los procedimientos comunes que cada comunidad autónoma ejecutará en su ámbito competencial, con el fin de facilitar su difusión y conocimiento.
En su tramitación se ha obtenido el informe preceptivo de la Agencia Española de Protección de Datos, y han sido oídos los sectores afectados y consultadas las comunidades autónomas.
En su virtud y previa aprobación de la Vicepresidenta Primera del Gobierno y Ministra de la Presidencia, dispongo:
Esta orden tiene por objeto publicar las Directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano, que figuran como anexo a la misma, y que han sido adoptadas por el Comité de Coordinación de Estudios Posautorización, en el ejercicio de sus funciones.
Esta orden tiene carácter de legislación de productos farmacéuticos a los efectos previstos en el artículo 149.1.16ª de la Constitución.
La presente orden entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado».
Madrid, 16 de diciembre de 2009.–La Ministra de Sanidad y Política Social, Trinidad Jiménez García-Herrera.
Los estudios posautorización se consideran necesarios para la obtención de un conocimiento que los ensayos clínicos controlados realizados durante el desarrollo clínico de los medicamentos no aportan. Dicho conocimiento es fundamental para orientar la práctica clínica y favorecer un uso racional de los medicamentos.
El Real Decreto 711/2002, de 19 de julio, por el que se regulaba la farmacovigilancia de medicamentos de uso humano y la Circular 15/2002 de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) constituyeron un primer paso en el control de estos estudios y en la garantía de los fines de los mismos. En este real decreto se prohibía expresamente la utilización de estudios posautorización como este tipo de prácticas promocionales encubiertas y se establecía que las administraciones sanitarias, en el ámbito de sus competencias, debían regular las condiciones en las que se realizarían dichos estudios, al objeto de favorecer los que tuvieran verdadero interés científico e impedir aquellos con fines promocionales. Este mandato se desarrolló a través de normativas específicas por cada una de las comunidades autónomas (CC.AA.) con competencias en ejecución de la legislación sobre productos farmacéuticos, que tomaron como referencia el anexo VI de la citada Circular 15/2002. A la AEMPS le asignaba dicho real decreto la responsabilidad de mantener un registro común para todo el Estado de todas las propuestas de estudio que se solicitaran. Dicho registro (base de datos GESTO) está operativo desde 2003 y es accesible para todas las CC.AA.
La citada regulación, y en particular, la autorización previa de los estudios posautorización de seguimiento prospectivo por parte de las CC.AA., ha permitido mejorar el control y la calidad de estos estudios. Superada esta etapa, por tanto, procede actualizar la normativa de referencia con el objeto de diferenciar con claridad los distintos tipos de estudios posautorización, e introducir mecanismos que permitan reducir la carga burocrática de aquellos que sean de gran interés para la salud pública, como son, entre otros, los requeridos por las autoridades sanitarias por motivos de seguridad o los financiados con fondos públicos. Las bases legales para esta diferenciación fueron introducidas por el nuevo el Real Decreto 1344/2007, de 11 de octubre, por el que se regula la farmacovigilancia de medicamentos de uso humano.
Un elemento clave que ha introducido el Real Decreto 1344/2007 de 11 de octubre ha sido el mandato de crear un Comité de Coordinación de Estudios Posautorización, formado por representantes de todas las CC.AA. y de la AEMPS, como foro de debate en el que se acuerden las directrices de los procedimientos comunes que tendrán que aplicar las CC.AA. en su ámbito de competencia. Dicho Comité fue constituido el 24 de septiembre de 2008. Las presentes directrices son el fruto de las deliberaciones de dicho Comité y reflejan por tanto la participación de todas las CC.AA. y la AEMPS en el proceso de toma de decisiones y el común acuerdo sobre su redactado.
Para la elaboración de estas directrices se ha tenido en cuenta, además, lo estipulado en el Capítulo I.7 sobre Estudios Posautorización promovidos por compañías farmacéuticas del Volumen 9A de las Normas sobre Medicamentos de la Unión Europea.
Las directrices se aplican a todos los estudios posautorización de tipo observacional que se realicen con medicamentos de uso humano. No obstante, se establecen cuatro procedimientos diferentes en función del tipo de estudio posautorización observacional de que se trate.
Cuando un estudio posautorización tenga carácter de ensayo clínico, de conformidad con la definición que figura en el artículo 58 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de medicamentos y productos sanitarios, no se regirá por lo dispuesto en las presentes directrices sino que le resultará de aplicación la normativa específica sobre ensayos clínicos, en particular el Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos.
Datos de carácter personal: (Real Decreto 1720/2006, de 21 de diciembre, por el que se aprueba el Reglamento de desarrollo de la Ley Orgánica 15/1999, de 13 de diciembre, de protección de datos de carácter personal): Cualquier información numérica, alfabética, gráfica, fotográfica, acústica o de cualquier otro tipo concerniente a personas físicas identificadas o identificables.
Procedimiento de disociación: (Real Decreto 1720/2006, de 21 de diciembre): Todo tratamiento de datos personales que permita la obtención de datos disociados.
Estudio posautorización: (Real Decreto 1344/2007, de 11 de octubre): Cualquier estudio clínico o epidemiológico realizado durante la comercialización de un medicamento según las condiciones autorizadas en su ficha técnica, o bien en condiciones normales de uso, en el que el medicamento o los medicamentos de interés son el factor de exposición fundamental investigado. Este estudio podrá adoptar la forma de un ensayo clínico o un estudio observacional.
Estudio posautorización de seguridad: (Real Decreto 1344/2007, de 11 de octubre): Estudio farmacoepidemiológico o ensayo clínico efectuado de conformidad con las disposiciones de la autorización de comercialización y realizado con el propósito de identificar, caracterizar o cuantificar los riesgos asociados a los medicamentos autorizados.
Estudio observacional: (Ley 29/2006, de 26 de julio): Estudio en el que los medicamentos se prescriben de la manera habitual, de acuerdo con las condiciones establecidas en la autorización. La asignación de un paciente a una estrategia terapéutica concreta no estará decidida de antemano por el protocolo de un ensayo, sino que estará determinada por la práctica habitual de la medicina, y la decisión de prescribir un medicamento determinado estará claramente disociada de la decisión de incluir al paciente en el estudio. No se aplicará a los pacientes ninguna intervención, ya sea diagnóstica o de seguimiento, que no sea la habitual de la práctica clínica, y se utilizarán métodos epidemiológicos para el análisis de los datos recogidos.
En el Capítulo I.7 del Volumen 9A, se aclara que «las entrevistas, los cuestionarios y las muestras de sangre se pueden considerar como práctica clínica habitual». En todo caso, para el manejo de muestras biológicas se seguirá lo dispuesto en la Ley 14/2007, de 3 de julio, de Investigación biomédica.
Estudio posautorización de tipo observacional: Estudio epidemiológico que cumple las condiciones de ser posautorización y observacional.
Estudio posautorización observacional de seguimiento: Todo aquel estudio posautorización de tipo observacional en el que los pacientes son seleccionados por su exposición a un determinado medicamento y son después seguidos durante un período de tiempo suficiente en relación con el acontecimiento de interés. Se consideran prospectivos cuando el periodo de estudio es posterior al inicio de la investigación y retrospectivos cuando el periodo de estudio es todo él anterior al inicio de la investigación.
Fuente de información: Origen de los datos que se utilizan para la realización del estudio.
El objetivo de los estudios posautorización es generar información adicional sobre los efectos de los medicamentos, así como las características relacionadas con su utilización, en las condiciones autorizadas en su ficha técnica o bien en condiciones normales de uso, con el fin de completar la información obtenida durante las fases I, II y III y contribuir a su mejor utilización.
Más concretamente, los estudios posautorización pueden realizarse con alguno de los siguientes fines:
Determinar la efectividad de los fármacos, es decir sus efectos beneficiosos en las condiciones de la práctica clínica habitual, así como los factores modificadores de la misma, tales como el incumplimiento terapéutico, la polimedicación, la gravedad de la enfermedad, presencia de enfermedades concomitantes, grupos especiales (ancianos, niños, etc.), factores genéticos o factores relacionados con el estilo de vida.
Identificar y cuantificar los efectos adversos del medicamento, en especial los no conocidos antes de la autorización, e identificar los posibles factores de riesgo o modificadores de efecto (características demográficas, medicación concomitante, factores genéticos, etc.). Con frecuencia, esto solo podrá estudiarse con precisión en grupos amplios de población y durante tiempos de observación prolongados.
Obtener nueva información sobre los patrones de utilización de medicamentos (dosis, duración del tratamiento, utilización apropiada).
Evaluar la eficiencia de los medicamentos, es decir la relación entre los resultados sanitarios y los recursos utilizados, utilizando para ello análisis farmacoeconómicos, tales como los de coste-efectividad, coste-utilidad, coste-beneficio o comparación de costes.
Conocer los efectos de los medicamentos desde la perspectiva de los pacientes (calidad de vida, satisfacción con los tratamientos recibidos, etc.).
Para que un estudio sea éticamente justificable debe estar bien diseñado y cumplir con los principios éticos básicos contenidos en la Declaración de Helsinki de la Asociación Médica Mundial sobre principios éticos para las investigaciones médicas en seres humanos, y en sus posteriores revisiones.
Todos los estudios posautorización de tipo observacional deben ser sometidos a la consideración de un Comité Ético de Investigación Clínica (CEIC) (o Comité de Ética de la Investigación, en su caso) acreditado, con la excepción de aquellos estudios que se realicen mediante la utilización de información ya existente que no contengan datos de carácter personal.
En los estudios que requieran entrevistar al sujeto o en aquellos en los que, utilizando otras fuentes de información, no sea posible adoptar un procedimiento de disociación seguro que garantice que la información que se maneja no contenga datos de carácter personal, se solicitará el consentimiento informado de los sujetos, el cual deberá ser otorgado por escrito, de acuerdo con la normativa vigente.
Cuando los sujetos sean menores o incapaces se solicitará el consentimiento informado de los tutores legales.
El promotor y los investigadores del estudio deben garantizar la confidencialidad de los datos de los sujetos y velar porque se cumpla en todo momento con lo establecido por la Ley Orgánica 15/1999, de 13 de diciembre, de protección de datos de carácter personal y Real Decreto 1720/2007, de 21 de diciembre.
Los estudios posautorización de tipo observacional están exentos de la obligatoriedad de suscripción de un seguro.
Cuando se trate de un estudio posautorización observacional de seguimiento prospectivo, el promotor y el investigador principal deberán expresar específicamente en el protocolo los procedimientos que se emplearán para garantizar que la realización del estudio no modificará los hábitos de prescripción del médico, o de dispensación del farmacéutico (en caso de medicamentos que no requieran prescripción). La prescripción del medicamento habrá de seguir los cauces habituales. Si se considera necesario un suministro del medicamento diferente al habitual, deberá justificarse apropiadamente en el protocolo.
Los investigadores deberán asegurarse de que su participación en el estudio no interfiere con sus cometidos asistenciales.
Los investigadores podrán recibir una compensación proporcional al tiempo y responsabilidades adicionales dedicados al estudio, sin perjuicio de las normas internas de las entidades empleadoras de los investigadores relativas a esta cuestión. La percepción económica habrá de ser, en todo caso, explícita y transparente y deberá ser puesto en conocimiento del CEIC que evalúe el estudio. La participación en el estudio de los investigadores habrá de ser libre, voluntaria e independiente.
6.1 Identificación de los responsables del estudio.
En todos los estudios deben identificarse las figuras del promotor y el investigador principal o coordinador como responsables últimos de la investigación.
Es promotor de un estudio posautorización toda aquella persona física o jurídica que tiene interés en su realización y asume las siguientes obligaciones:
a) Firmar con el investigador coordinador el protocolo y cualquier modificación del mismo.
b) Suministrar a los investigadores el protocolo y la ficha técnica de los medicamentos a estudiar.
c) Remitir el protocolo al CEIC (o Comité de Ética de la Investigación, en su caso).
d) Solicitar la autorización de la Administración, cuando proceda, y presentar la documentación correspondiente.
e) Presentar los informes de seguimiento y final, en los plazos establecidos y comunicar, en su caso, la interrupción y las razones de la misma.
f) Entregar copia del protocolo y de los documentos que acrediten el seguimiento de los procedimientos establecidos en las presentes directrices a los responsables de las entidades proveedoras de servicios de atención a la salud donde se vaya a realizar el estudio.
g) Aplicar un control de calidad en la obtención y el manejo de datos para asegurar que los datos son fiables.
h) Comunicar las sospechas de reacciones adversas graves que surjan a lo largo del estudio al punto de contacto designado por el órgano competente en materia de farmacovigilancia de la comunidad autónoma donde ejerza su actividad el profesional sanitario que comunicó el caso.
i) Identificar las fuentes de financiación del estudio
j) Firmar, en su caso, el contrato con la entidad competente
k) Hacer públicos los resultados del estudio, a ser posible, a través de una revista científica.
El promotor podrá designar a un monitor como vínculo entre él y los investigadores. Su principal obligación es asegurarse de que el estudio se está realizando conforme a lo exigido en el protocolo. Para ello, podrá realizar cuantas comprobaciones considere necesarias.
Los profesionales sanitarios que contribuyan al estudio recogiendo información serán considerados como investigadores. Son obligaciones de éstos:
a) Firmar un compromiso en el que se reconocen como investigadores del estudio y afirman que conocen el protocolo y cualquier modificación del mismo, y están de acuerdo con él en todos sus términos.
b) Informar a los sujetos de investigación y obtener su consentimiento.
c) Recoger, registrar y notificar los datos de forma correcta respondiendo de su actualización y calidad ante las auditorías oportunas.
d) Notificar al promotor los acontecimientos adversos según se establezca en el protocolo.
e) Respetar la confidencialidad de los datos del sujeto.
f) Facilitar las visitas de monitorización del monitor, las auditorías del promotor y las inspecciones de las autoridades sanitarias.
g) Saber responder sobre los objetivos, metodología básica y significado de los resultados del estudio ante la comunidad científica y profesional.
Es investigador coordinador el profesional sanitario que dirige científicamente el estudio. Son obligaciones específicas del investigador coordinador las enumeradas anteriormente más las siguientes:
a) Firmar el protocolo y cualquier modificación del mismo junto con el promotor.
b) Co-responsabilizarse con el promotor de la elaboración de los informes de seguimiento y finales.
c) Contribuir a difundir los resultados del estudio, en colaboración con el promotor.
6.2 Elementos del Protocolo.
Siempre que sea posible, los protocolos deberán ajustarse, de forma general, a la siguiente estructura (basada en Capítulo I.7. Volumen 9A, Tabla I.7.B):
A. Título descriptivo y versión del protocolo.
B. Responsable del estudio (nombres, títulos, grados, especialidad, lugar de trabajo y direcciones de todos los responsables, incluyendo investigador coordinador, otros investigadores y monitor cuando proceda).
C. Promotor (nombre y dirección. En su caso, titular de la autorización de comercialización).
D. Resumen:
1. Identificación del promotor y dirección.
2. Título del estudio.
3. Código del protocolo (según normas oficiales de codificación).
4. Investigador principal y dirección.
5. Tipo de centros donde se prevé realizar el estudio.
6. CEIC que lo evalúa.
7. Objetivo principal.
8. Diseño.
9. Enfermedad o trastorno en estudio.
10. Datos de los medicamentos objeto de estudio.
11. Población en estudio y número total de sujetos.
12. Calendario.
13. Fuente de financiación.
E. Plan de trabajo (tareas, hitos y cronología del estudio).
F. Objetivos generales y específicos. Fundamentos.
G. Revisión crítica de la literatura.
H. Métodos:
1. Diseño y justificación.
2. Población de estudio.
3. Fuente de información.
4. Definición operativa de variables de resultado, exposición y otras.
5. Tamaño de la muestra previsto y bases para su determinación.
6. Métodos para la obtención de los datos.
7. Manejo de los datos.
8. Análisis de los datos.
9. Control de calidad.
10. Limitaciones del diseño, de la fuente de información y de los métodos de análisis.
I. Aspectos éticos/protección de los sujetos participantes:
1. Evaluación beneficio-riesgo para los sujetos de investigación.
2. Consideraciones sobre información a los sujetos y consentimiento informado..
3. Confidencialidad de los datos.
4. Interferencia con los hábitos de prescripción del médico.
J. Manejo y comunicación de reacciones adversas.
K. Planes para la difusión de los resultados.
L. Recursos para la realización del estudio y asignación de tareas. Forma de suministro del medicamento. Financiación.
M. Bibliografía.
N. Modificaciones del protocolo.
Ñ. Consideraciones prácticas:
1. Informes de seguimiento y final.
2. Difusión de los resultados
O. Anexos (al menos los siguientes):
1. Anexo 1: Cuaderno de recogida de datos.
2. Anexo 2: Compromiso del investigador coordinador.
3. Anexo 3: Conformidad del CEIC.
4. Anexo 4: Ficha Técnica del medicamento investigado.
5. Anexo 5: Hoja de información a los sujetos.
6. Anexo 6: Formulario de consentimiento informado.
7. Anexo 7: Memoria económica.
6.3 Identificación de estudio.
Sin perjuicio del código interno del protocolo de cada promotor, todos los protocolos de estudios posautorización deben identificarse con un código de 12 dígitos siguiendo las siguientes normas de codificación:
Posiciones 1-3: tres primeras letras del promotor
Posiciones 4-6: tres primeras letras del principio activo de interés (uno de ellos si hay más de uno)
Posiciones 7-10: año en curso
Posiciones 11-12: Número secuencial de 2 dígitos
Ejemplo: El promotor PANDORA desea hacer un estudio con el principio activo ACTIVINA, en el año 2002 siendo el cuarto que hace este año con dicho principio activo.
Código: PAN-ACT-2002-04
Solo se consignará un código por protocolo. Por ejemplo, si un mismo estudio se va a realizar en 4 CC.AA., en todas ellas se identificará por el mismo código.
En el Real Decreto 1344/2007, de 11 de octubre, en su artículo 19.3 establece que «la Agencia mantendrá un registro de las propuestas de estudio posautorización de tipo observacional, al que tendrán acceso los órganos competentes de las comunidades autónomas, e informará a cada promotor sobre los procedimientos a seguir en cada caso. A tal efecto, el promotor del estudio deberá remitir a la Agencia Española de Medicamentos y Productos Sanitarios el protocolo del estudio».
De acuerdo con esto, la AEMPS realizará una clasificación previa de todos los estudios clínicos o epidemiológicos no aleatorizados, que se realicen con seres humanos o con registros médicos y que tengan uno o varios medicamentos como exposición de interés en el plazo máximo de 30 días naturales desde su recepción. Para ello el promotor deberá remitir a la AEMPS el protocolo completo o un resumen amplio del mismo, donde queden perfectamente explícitos los objetivos y el diseño del estudio, indicando que el destinatario es la División de Farmacoepidemiología y Farmacovigilancia de la AEMPS. Es relevante que en esta comunicación se indique claramente que se remite para su CLASIFICACIÓN, así como la dirección postal y electrónica y el teléfono de contacto del promotor.
Se podrá presentar el protocolo en paralelo a la AEMPS para clasificación y al CEIC para evaluación. Una vez recibido el protocolo o su resumen amplio, la AEMPS remitirá al solicitante la resolución correspondiente indicando la clasificación del estudio y la ruta administrativa que el mismo deberá seguir (ver cuadro I). Para los casos de dudosa clasificación la AEMPS solicitará formalmente un informe al Comité de Coordinación de Estudios Posautorización. En dicho caso se producirá una suspensión del plazo por el tiempo que medie entre la petición de dicho informe, que será comunicada al interesado, y la recepción del mismo. Este plazo de suspensión no podrá exceder en ningún caso de tres meses.
Toda aquella documentación que se remita a la AEMPS para la clasificación de un estudio, no tendrá que ser reenviada en el momento de la solicitud de autorización y/o registro del mismo.
La AEMPS mantendrá una base de datos con la información relativa a todos los estudios posautorización que se remitan para su registro, a la que tendrán acceso para consulta y, en su caso, carga de la información correspondiente, los órganos competentes de las CC.AA. En dicha base de datos constarán los aspectos administrativos y metodológicos fundamentales de los estudios.
Todos aquellos estudios que se consideren ensayos clínicos deberán seguir la normativa específica.
En el Cuadro I se resumen de forma esquemática los procedimientos administrativos que se detallan a continuación para cada tipo de estudio.
7.1 Procedimiento para la autorización de estudios posautorización de tipo observacional que sean una condición establecida en el momento de la autorización de un medicamento, o bien constituya una exigencia de la autoridad competente para aclarar cuestiones relativas a la seguridad del medicamento, o forme parte del plan de gestión de riesgos.
Este tipo de estudios se abreviarán como EPA-LA.
La normativa aplicable es el Real Decreto 1344/2007, de 11 de octubre, donde se recoge en el artículo 19.5 lo siguiente:
«Cuando la realización de un estudio posautorización de tipo observacional sea una condición establecida en el momento de la autorización de un medicamento, o bien constituya una exigencia de la autoridad competente para aclarar cuestiones relativas a la seguridad del medicamento, o forme parte del plan de gestión de riesgos que debe llevar a cabo el titular requerirá únicamente la autorización de la Agencia Española de Medicamentos y Productos Sanitarios, según los procedimientos que se establezcan al efecto. La Agencia Española de Medicamentos y Productos Sanitarios informará de estos estudios a las comunidades autónomas donde se vayan a realizar y los incluirá en el registro referido en el apartado 3».
Para todos los estudios clasificados como EPA-LA, el promotor deberá presentar la documentación siguiente, indicando como destinatario la División de Farmacoepidemiología y Farmacovigilancia de la AEMPS:
Carta de presentación dirigida a la División de Farmacoepidemiología y Farmacovigilancia de la AEMPS donde se solicite la autorización del estudio e indique la dirección y contacto del solicitante y la relación de documentos que se incluyen. En el caso de que el promotor no sea quien presente la documentación, se deberá incluir en la misma un documento que indique las responsabilidades delegadas por el promotor a la persona o empresa que actúa en su nombre.
Formulario resumen del protocolo (disponible en: www.aemps.es).
Protocolo completo, incluidos los anexos y cuaderno de recogida de datos, y donde conste el número de pacientes que se pretenden incluir en España.
Dictamen favorable del estudio por un CEIC acreditado en España.
Acreditación documental de que el estudio ha sido requerido por la autoridad competente, o que forme parte del plan de gestión de riesgos del producto.
Acreditación documental de la revisión por parte de la autoridad sanitaria competente que instó la realización del estudio.
Listado de CC.AA y centros sanitarios en los que se pretende realizar el estudio, así como los investigadores participantes.
Si el estudio se pretende realizar en otros países, situación del mismo en éstos.
La AEMPS evaluará la pertinencia del estudio y resolverá favorable o desfavorablemente el mismo en el plazo máximo de 60 días naturales desde su recepción. Si pasado este plazo, la AEMPS no ha emitido informe desfavorable se dará por aprobado el estudio.
En el caso de que fuera precisa la aportación de cualquier documentación o información adicional a efectos de la autorización del estudio, y una vez requerida la misma, se podrá suspender el plazo para la resolución del procedimiento de acuerdo con lo establecido en el artículo 42.5 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, siendo en este caso notificada dicha suspensión al interesado en la comunicación del requerimiento.
Una vez aprobado el estudio por la AEMPS, deberá contratarse la realización con los centros sanitarios donde se vaya a realizar, siguiendo los requisitos que cada comunidad autónoma establezca, lo que incluirá necesariamente la resolución de la AEMPS por la que se autoriza el estudio.
Para los estudios que formen parte del plan de gestión de riesgos, es necesario que se solicite su autorización de manera independiente a la presentación en la AEMPS del plan de gestión de riesgos.
La AEMPS mantendrá informados a los órganos competentes de las CCAA. sobre la autorización de estos estudios, a través de la aplicación informática de registro de estudios posautorización u otro procedimiento alternativo que se acuerde.
No se podrán realizar estudios EPA-LA sin la preceptiva autorización de la AEMPS. El incumplimiento de lo anteriormente expuesto dará lugar a la incoación del correspondiente expediente sancionador.
7.2 Procedimiento simplificado para la autorización de estudios posautorización de tipo observacional de seguimiento prospectivo promovidos por las Administraciones sanitarias o financiados con fondos públicos.
Este tipo de estudios se abreviarán como EPA-AS
De acuerdo con el artículo 19.6 del Real Decreto 1344/2007, de 11 de octubre, «cuando se trate de estudios promovidos por las Administraciones sanitarias o financiados con fondos públicos, se establecerán procedimientos simplificados al objeto de facilitar su realización y que se acordarán en el Comité de Coordinación de Estudios Posautorización».
El promotor de un estudio posautorización de seguimiento prospectivo que responda a los criterios mencionados, enviará el protocolo del estudio, junto con el formulario resumen del mismo (disponible en www.aemps.es) y el dictamen favorable del CEIC, a la Secretaría del Comité de Coordinación de Estudios Posautorización (División de Farmacoepidemiología y Farmacovigilancia de la AEMPS).
La Secretaría del Comité distribuirá el protocolo del estudio a sus miembros, junto con un informe en el que consten las razones de su inclusión en la categoría de estudios promovidos por las Administraciones sanitarias o financiados con fondos públicos. El Comité emitirá informe sobre la solicitud de autorización del estudio, correspondiendo a la AEMPS, de acuerdo con el pronunciamiento previo del Comité, emitir la resolución correspondiente. Dicha resolución se emitirá en un plazo máximo de 30 días naturales a contar desde la solicitud de autorización. Si pasado este plazo, la AEMPS no ha emitido informe favorable se dará por autorizado el estudio.
En el caso de que fuera precisa la aportación de cualquier documentación o información adicional a efectos de la autorización del estudio, y una vez requerida la misma, se podrá suspender el plazo para la resolución del procedimiento de acuerdo con lo establecido en el artículo 42.5 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, siendo en este caso notificada dicha suspensión al interesado en la comunicación del requerimiento.
El promotor informará a los responsables de las entidades proveedoras de servicios sanitarios donde prevea llevarse a cabo el estudio, o a los organismos que la comunidad autónoma haya designado para este fin, y les entregará copia del protocolo y de los documentos que acrediten su aprobación. La gestión y formalización del contrato estará sujeta a los requisitos específicos de cada comunidad autónoma.
7.3 Procedimiento para la autorización de estudios posautorización de tipo observacional de seguimiento prospectivo con medicamentos, no incluidos en los epígrafes anteriores (7.1 y 7.2).
Este tipo de estudios se abreviarán como EPA-SP.
Una vez clasificado el estudio como EPA-SP por la AEMPS, el promotor presentará el protocolo del estudio, junto con el material informativo que se prevea enviar a los profesionales sanitarios, a los órganos competentes de las CC.AA. donde se vaya a realizar el estudio y simultáneamente a la AEMPS, una vez obtenida la conformidad de un CEIC acreditado, junto con el formulario resumen del estudio (disponible en www.aemps.es). El promotor hará constar en el protocolo si el estudio se ha presentado con anterioridad a algún otro CEIC e informará del resultado de su evaluación.
El promotor deberá presentar en las CC.AA. donde se pretenda realizar el estudio la siguiente documentación:
Carta de presentación dirigida a los responsables de esta materia en la comunidad autónoma en la que se solicite la autorización del estudio e indique la dirección y contacto del solicitante y la relación de documentos que se incluyen. En el caso de que el promotor no sea quien presente la documentación, se deberá incluir en la misma un documento que indique las responsabilidades delegadas por el promotor a la persona o empresa que actúa en su nombre.
Formulario resumen del protocolo (disponible en: www.aemps.es).
Resolución de la AEMPS sobre la clasificación del estudio
Protocolo completo incluidos los anexos, y donde conste el número de pacientes que se pretenden incluir en España, desglosado por comunidad autónoma.
Dictamen favorable del estudio por un CEIC acreditado en España.
Listado de centros sanitarios donde se pretende realizar el estudio, desglosado por comunidad autónoma
Listado de investigadores participantes en la comunidad autónoma.
Si el estudio se pretende realizar en otros países, situación del mismo en éstos
Documento acreditativo de haber satisfecho las tasas correspondientes, en aquellas CC.AA. donde se exijan.
Los órganos competentes de las CC.AA. evaluarán la pertinencia del estudio y resolverán favorable o desfavorablemente el mismo en el plazo máximo de 90 días naturales desde su recepción. Si pasado este plazo la comunidad autónoma correspondiente no ha emitido informe desfavorable se dará por aprobado el estudio en el ámbito de dicha comunidad autónoma.
En el caso de que fuera precisa la aportación de cualquier documentación o información adicional a efectos de la autorización del estudio, y una vez requerida la misma, se podrá suspender el plazo para la resolución del procedimiento de acuerdo con lo establecido en el artículo 42.5 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, siendo en este caso notificada dicha suspensión al interesado en la comunicación del requerimiento.
La comunidad autónoma informará de la resolución a la AEMPS (normalmente a través de la aplicación informática), y al promotor.
El promotor informará a los responsables de las entidades proveedoras de servicios sanitarios donde prevea llevarse a cabo el estudio, o a los organismos que la comunidad autónoma haya designado para este fin, y les entregará copia del protocolo y de los documentos que acrediten su aprobación. La gestión y formalización del contrato estará sujeta a los requisitos específicos de cada comunidad autónoma.
Para la evaluación de los protocolos de los estudios y la revisión de los informes de seguimiento y finales, las CC.AA. podrán solicitar la colaboración de expertos, entre los que se encontrarán los técnicos correspondientes del Sistema Español de Farmacovigilancia de medicamentos de uso humano.
No se podrán realizar estudios EPA-SP sin la preceptiva autorización de los órganos competentes de las comunidades autónomas involucradas. El incumplimiento de lo anteriormente expuesto dará lugar a la incoación del correspondiente expediente sancionador.
7.4 Procedimiento a seguir para otros estudios posautorización de tipo observacional.
Este tipo de estudios se abreviarán como EPA-OD.
En este grupo se encuentran los estudios posautorización que no hayan sido categorizados como EPA-LA y respondan a diseños diferentes al de «seguimiento prospectivo», por ejemplo, estudios de casos y controles, estudios transversales o estudios de cohorte retrospectivos. Una vez clasificado el estudio como EPA-OD por la AEMPS, el promotor deberá remitir a la AEMPS el protocolo junto con la aprobación del mismo por un CEIC acreditado en España y el formulario resumen del protocolo (disponible en www.aemps.es). Si para la clasificación del estudio se presenta esta documentación, no requerirá ningún trámite adicional. La AEMPS procederá a registrar oportunamente el estudio e informará a los órganos competentes de las CC.AA. En el caso de que el promotor no sea quien presente la documentación, se deberá incluir en la misma un documento que indique las responsabilidades delegadas por el promotor a la persona o empresa que actúa en su nombre.
Los estudios EPA-OD no requieren autorización por parte de la AEMPS ni de los órganos competentes de las CC.AA. donde se vayan a realizar, si bien el promotor tendrá que informar a los responsables de las entidades proveedoras de servicios sanitarios donde se lleve a cabo el estudio y les entregará copia del protocolo y de los documentos que acrediten la aprobación por parte del CEIC y la clasificación de la AEMPS. Asimismo estos documentos se entregarán a los órganos competentes de las CC.AA., cuando sea requerido. La gestión y formalización del contrato estará sujeta a los requisitos específicos de cada comunidad autónoma.
7.5 Procedimiento para la realización de estudios observacionales que no sean posautorización.
Estos estudios se abreviarán como No-EPA.
Se trata de aquellos estudios en los que el factor de exposición fundamental investigado no son medicamentos. En caso de que se recoja información sobre medicamentos, el protocolo tendrá que ser presentado a la AEMPS para su clasificación. La AEMPS procederá a registrar oportunamente el estudio e informará a los órganos competentes de las CC.AA.
Los estudios No-EPA no requieren autorización por parte de la AEMPS ni de los órganos competentes de las CC.AA. donde se vayan a realizar, pero sí es necesario presentarlo a un CEIC acreditado y obtener su dictamen favorable. El promotor tendrá que informar a los responsables de las entidades proveedoras de servicios sanitarios donde se lleve a cabo el estudio y les entregará copia del protocolo y de los documentos que acrediten la aprobación por parte del CEIC y, en su caso, la clasificación de la AEMPS. Asimismo estos documentos se entregarán a los órganos competentes de las CC.AA., cuando sea requerido. La gestión y formalización del contrato estará sujeta a los requisitos específicos de cada comunidad autónoma.
Todos los estudios posautorización requerirán para su inicio el oportuno contrato del promotor con los responsables de las entidades proveedoras de servicios sanitarios donde prevea llevarse a cabo el estudio o el visto bueno del director del centro conforme a los procedimientos específicos que se manejen en cada centro.
El promotor comunicará la fecha efectiva de comienzo del estudio a los órganos competentes de las CC.AA. donde se prevea realizar y a la AEMPS, a menos que ya venga indicado en el propio protocolo.
8.1 Enmiendas al protocolo
Cuando la enmienda afecte a aspectos fundamentales del protocolo del estudio (en particular a los apartados de objetivos, métodos, aspectos éticos – ver 6.2), se someterá de nuevo a la evaluación del CEIC que informó favorablemente sobre el mismo y se solicitará autorización administrativa para dicha enmienda a quien corresponda en función del tipo de estudio. Para el resto de enmiendas bastará con que se informe de las modificaciones efectuadas, justificando por qué dicha enmienda no afecta a aspectos fundamentales del protocolo del estudio.
En caso de duda sobre la consideración de la enmienda, se puede remitir consulta a la AEMPS a través de la dirección de correo electrónico farmacoepi@aemps.es.
8.2 Informes de seguimiento y finales.
Para los estudios EPA-LA, EPA-SP y EPA-AS que sean considerados como de seguimiento prospectivo el promotor deberá enviar un informe de seguimiento anual, o antes si así se solicita. Asimismo, el promotor informará de forma inmediata sobre cualquier incidencia relevante (interrupción, problema grave de seguridad, etc.) que pueda producirse en el transcurso del estudio. Todas estas comunicaciones se presentarán a los órganos competentes de las CC.AA. involucradas y a la AEMPS.
Para todos los estudios posautorización, el promotor deberá remitir un informe final entre los tres y seis meses después de la finalización del estudio, a la AEMPS y a los órganos competentes de las CC.AA. donde se realizó.
8.3 Comunicación de sospechas de reacciones adversas
Para los estudios catalogados como EPA-SP, EPA-AS y para los EPA-LA que sean considerados como de seguimiento prospectivo, las sospechas de reacciones adversas graves que se detecten durante el transcurso del estudio se notificarán al punto de contacto designado por el órgano competente en materia de farmacovigilancia de la comunidad autónoma donde ejerza su actividad el profesional sanitario que notifique el caso, en el plazo máximo de 15 días naturales desde que se tuvo conocimiento de la sospecha de reacción adversa. Esta notificación se realizará obligatoriamente de forma electrónica si el promotor es una compañía farmacéutica, por parte del responsable de farmacovigilancia siguiendo las instrucciones publicadas la AEMPS. La AEMPS pondrá a disposición del titular de la autorización de comercialización dentro de los 15 días naturales siguientes a su recepción, las notificaciones sobre sospechas de reacciones adversas graves que se detecten en el transcurso de un EPA, haciendo referencia al código del estudio.
En el caso de que el promotor sea un grupo de profesionales (vgr. sociedades científicas) podrá optar por una de la dos siguientes opciones: 1) tarjeta amarilla al centro autonómico de farmacovigilancia correspondiente, indicando en observaciones el nombre y el código del estudio del cual proviene; o 2) utilizar la carga on-line a través del portal SINAEM de la AEMPS, siguiendo las instrucciones publicadas por la AEMPS (esta segunda opción es la recomendada).
Si el promotor es un profesional sanitario notificará las sospechas de reacciones adversas a través de tarjeta amarilla al centro autonómico de farmacovigilancia correspondiente, indicando igualmente el nombre y el código del estudio del cual proviene.
De acuerdo con el Capítulo I.7 del Volumen 9A, aquellos estudios en los que no sea posible o no sea apropiado hacer una evaluación individual de la relación de causalidad entre los acontecimientos clínicos y los medicamentos de interés, la notificación individual de sospechas de reacciones adversas no será necesaria. Los estudios etiquetados como EPA-OD, o EPA-LA que no sean de seguimiento prospectivo, entran dentro de esta categoría y para ellos no se precisará la notificación expeditiva de sospechas de reacciones adversas, a menos que se indique otra cosa por la AEMPS en el momento del registro del estudio.
Además de lo especificado en el párrafo anterior, cualquier problema de seguridad relevante que se detecte durante el transcurso del estudio será puesto en conocimiento de la AEMPS y de los órganos competentes de las CC.AA. involucradas, con independencia del diseño y catalogación del estudio.
8.4 Archivo de los documentos del estudio:
La documentación relativa al estudio posautorización constituye el archivo maestro del mismo y constará de los documentos esenciales que permitan la evaluación de la realización de un estudio posautorización y de la calidad de los datos obtenidos. Estos documentos deberán demostrar el cumplimiento por parte del investigador y del promotor de los requisitos establecidos para los estudios posautorización.
El archivo maestro del estudio posautorización proporcionará la base para las auditorías que pueda realizar el promotor a través de auditores independientes y para las inspecciones de las autoridades competentes.
El promotor y el investigador conservarán los documentos y materiales esenciales de cada estudio durante al menos cinco años tras la finalización del mismo, o durante un período más largo si así lo disponen otros requisitos aplicables.
Los documentos y materiales esenciales deberán archivarse de forma que se puedan poner fácilmente a disposición de las autoridades competentes, en caso de que los soliciten.
La historia clínica de cada paciente del estudio deberá ser custodiada con arreglo a los dispuesto en la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica, y la legislación autonómica aplicable, y conforme al período máximo establecido por el hospital, la institución o la consulta privada. Asimismo, se deberá contemplar lo dispuesto en la Ley Orgánica 15/1999, de 13 de diciembre, de protección de datos de carácter personal y el Real Decreto 1720/2007, de 21 de diciembre, por el que se aprueba el reglamento de desarrollo de dicha ley.
Todos los cambios en la titularidad de los datos, documentos y materiales deberán documentarse. El nuevo titular asumirá las responsabilidades de las tareas de archivo y conservación de los datos.
El promotor nombrará la persona de su organización responsable de los archivos y el acceso a los mismos deberá limitarse a las personas designadas.
Los soportes utilizados para la conservación de los documentos esenciales deberán garantizar que los documentos y materiales permanezcan completos y legibles durante el período de conservación previsto y que estén a disposición de las autoridades competentes en caso de que los soliciten. Cualquier modificación de los registros habrá de ser trazable, permitiendo conocer el dato inicial y el corregido, así como la fecha y firma del autor del cambio.
8.5 Inspecciones:
Las autoridades sanitarias competentes de las CC.AA., en el ámbito de sus competencias, verificarán el cumplimiento de las prescripciones legales relativas a los estudios posautorización que se realicen en España, a través de las correspondientes inspecciones y de acuerdo con los procedimientos que se establezcan.
Las inspecciones serán llevadas a cabo antes, durante o después de la realización del estudio por inspectores debidamente cualificados. Se podrán hacer inspecciones en los lugares relacionados con la realización del estudio y, entre otros, en el centro o centros sanitarios en los que se lleve a cabo el estudio, en cualquier laboratorio de análisis o centro de diagnóstico utilizado, en las instalaciones del promotor y/o de las organizaciones o empresas de investigación implicadas por contrato en la realización del estudio y al CEIC que lo evaluó.
CUADRO I: Rutas administrativas de los Estudios Posautorización
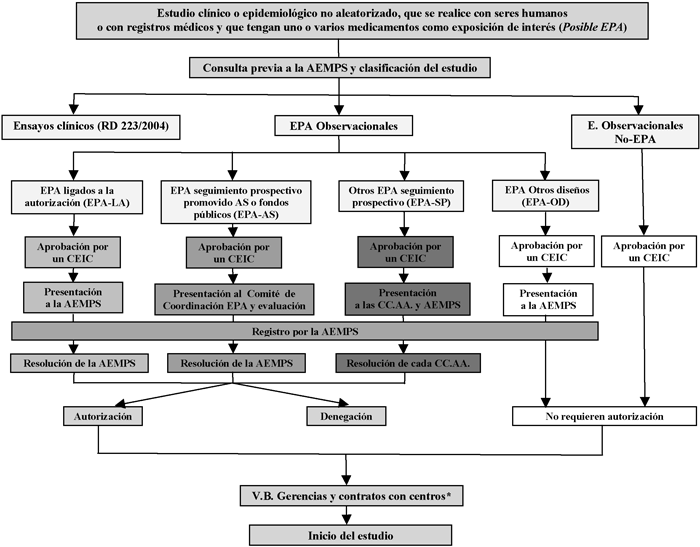
Abreviaturas y leyendas en el Cuadro I:
* En alguna comunidad autónoma este paso es previo a la autorización de la Administración Sanitaria.
EPA: Estudio posautorización.
EPA-LA: Estudios posautorización cuya realización tiene lugar a instancia de las autoridades reguladoras y ligada a la autorización de comercialización (AEMPS o Agencia Europea de Medicamentos). Se incluyen en esta vía administrativa tanto los estudios ligados a la autorización como los estudios de seguridad a requerimiento de las autoridades sanitarias y los incluidos en los planes de gestión de riesgos.
EPA-AS: Estudio posautorización promovido por las Administraciones Sanitarias (AS) o financiado con fondos públicos.
EPA-SP: Estudio posautorización de seguimiento prospectivo que no corresponde a ninguna de las dos categorías anteriores.
EPA-OD: Estudios posautorización con diseño diferente al de seguimiento prospectivo, por ejemplo, estudios transversales o retrospectivos.
No-EPA: Estudios en los que el factor de exposición fundamental investigado no es un medicamento; por ejemplo estudios de incidencia o de prevalencia de enfermedades, etc.
Este documento es de carácter informativo y no tiene valor jurídico.
Ayúdenos a mejorar: puede dirigir sus comentarios y sugerencias a nuestro Servicio de atención al ciudadano
Agència Estatal Butlletí Oficial de l'Estat
Avda. de Manoteras, 54 - 28050 Madrid





