Butlletí Oficial de l'Estat






Contingut no disponible en valencià
El Decreto dos mil quinientos diecinueve/mil novecientos setenta y cuatro, de nueve de agosto, regula la entrada en vigor, aplicación y desarrollo del Código Alimentario Español, respetando la vigencia de las normas especiales en materia alimentaria, en cuanto no sean expresamente modificadas para adaptarlas a la sistemática, principios básicos y características técnicas que el Código contiene.
Por otra parte, la sección segunda del capítulo XXIII del Código Alimentario Español, que abarca los artículos tres punto veintitrés punto veinticuatro al tres punto veintitrés punto veintinueve, ambos inclusive, presentaba algunos errores y lagunas que podían provocar dificultades en el momento de su puesta en vigor e interpretación.
En su virtud, a propuesta de los Ministros de la Gobernación, de Industria, de Agricultura y de Comercio, oída la Organización Sindical, de acuerdo con el informe favorable de la Comisión Interministerial para la Ordenación Alimentaria y previa deliberación del Consejo de Ministros en su reunión del día siete de marzo de mil novecientos setenta y cinco,
DISPONGO:
Se aprueba la adjunta Norma sobre la miel.
Esta Norma entrará en vigor al día siguiente de su publicación en el «Boletín Oficial del Estado».
Quedan derogadas cuantas disposiciones de igual o inferior rango se opongan al contenido de la presente Norma.
Así lo dispongo por el presente Decreto, dado en Madrid a siete de marzo de mil novecientos setenta y cinco.
FRANCISCO FRANCO
El Ministro de la Presidencia del Gobierno,
ANTONIO CARRO MARTINEZ
1. DESCRIPCION
1.2 Definición de miel.
Se entiende por miel la sustancia dulce producida por las abejas a partir del néctar de las flores o de exudaciones de otras partes vivas de las plantas o presente en ellas, que dichas abejas recogen, transforman y combinan con sustancias específicas y almacenan después en panales.
1.2 Características.
La miel se compone esencialmente de diferentes azúcares, predominantemente glucosa y fructosa. Además de glucosa y fructosa, la miel contiene proteínas, aminoácidos, enzimas, ácidos orgánicos, sustancias minerales, polen y otras sustancias, y puede contener sacarosa, maltosa, malecitosa y otros oligosacáridos (incluidas las dextrinas), así como vestigios de hongos, algas, levaduras y otras partículas sólidas, como consecuencia del proceso de obtención de la miel. El color de la miel varía desde casi incoloro a pardo oscuro o casi negro. Su consistencia puede ser fluida, viscosa o cristalizada total o parcialmente. El sabor y el aroma varían, pero, generalmente, posee los de las plantas de que procede.
1.3 Definiciones y: denominaciones subsidiarias.
1.3.1 Según su origen:
Miel de flores: Es la miel que procede principalmente de los néctares de las flores.
Miel de mielada; Es la miel que procede principalmente de exudaciones de las partes vivas de las plantas o presentes en ellas. Su color varía de pardo muy claro o verdoso a casi negro.
1.3.2. Según el procedimiento de obtención:
Miel en panal o en secciones: Es la miel depositada por las abejas en panales de reciente construcción y sin larvas, y vendida en panales enteros, no desoperculados o en secciones de panales.
Miel centrifugada: Es la miel que se obtiene mediante la centrifugación de los panales desoperculados, sin larvas.
Miel prensada: Es la miel obtenida mediante la compresión de los panales, sin larvas, con o sin aplicación de calor moderado.
2. FACTORES ESENCIALES. DE COMPOSICION Y CALIDAD
2.1 Criterios de composición.
2.1.1 Contenido aparente de azúcar reductor, calculado como azúcar invertido.
Miel de flores, cuando está rotulada como tal: No menos del 65 por 100.
Miel de mielada y mezclas de miel de mielada y miel de flores: No menos del 60 por 100.
2.1.2 Contenido de humedad: No más del 21 por 100.
Miel de brezo (Calluna): No más del 23 por 100.
2.1.3 Contenido aparente de sacarosa: No más del 5 por 100.
Miel de mielada y mezclas de miel de mielada y miel de flores: No más del 10 por 100.
2.1.4 Contenido de sólidos insolubles en agua: No más del 0,1 por 100.
Miel prensada: No más del 0,5 por 100.
2.1.5 Contenido de sustancias minerales (cenizas): De 0,1 a 0,6 por 100.
Miel de mielada y mezclas de miel de mielada y miel de flores: De 0,5 a 1,0 por 100.
2.1.6 Acidos: No más de 40 miliequivalentes de ácido por 1.000 gramos.
2.1.7 Contenido en dextrinas. Menos del 2 por 100, en la miel de flores, y hasta el 10 por 100, en la mielada.
2.1.8 Actividad de la diastasa y contenido de hidroximetilfurfural:
Indice de diastasa en la escala de Gothe, determinado después de la elaboración y mezcla: No menos de 8, siempre que el contenido de hidroximetilfurfural no sea mayor de 40 mg/Kg.
Mieles con un contenido bajo de enzimas naturales; por ejemplo, mieles de cítricos, contenido en diastasa en la escala de Gothe: No menos de 3, siempre que el contenido de hidroximetilfurfural no sea mayor de 15 mg/Kg.
2.2 Prohibiciones específicas.
2.2.1 La miel no deberá tener ningún sabor, aroma u olor que no sean los propios o genuinos.
2.2.2 La miel no deberá haber comenzado a fermentar ni ser efervescente.
2.2.3 La miel no deberá calentarse hasta tal grado que se inactiven totalmente, o en gran parte, las enzimas naturales que contiene (ver 2.1.7).
2.2.4 La acidez de la miel no deberá cambiarse artificialmente.
3. ADITIVOS ALIMENTARIOS Y ADICIONES
3.1 No se permite ninguno.
4. HIGIENE
4.1 El producto regulado por las disposiciones de esta norma se preparará y manipulará cumpliendo los requisitos aplicables a este producto, recogidos en el artículo 1.03.05 del Código Alimentario Español.
4.2 La miel, cuando se ponga a la venta al por menor o se utilice en cualquier producto destinado al consumo humano, deberá estar exenta de sustancias inorgánicas u orgánicas extrañas a su composición, tales como insectos, restos de insectos, larvas, granos de arena, contaminaciones microbianas o de residuos tóxicos (véase 2.1.4).
5. ETIQUETADO
El etiquetado de la miel se ajustará a la norma general para rotulación, etiquetado y publicidad, y específicamente deberá expresarse:
5.1. Nombre del producto.
5.1.1 Sólo podrán etiquetarse con el término «miel» los productos que satisfagan las disposiciones de la norma.
5.1.2 Ninguna miel podrá designarse con una de las denominaciones que figuran en el 1.3, a menos que se ajuste a la descripción correspondiente que figura en dicho apartado.
5.1.3 La miel deberá designarse según su procedencia floral o vegetal, si la parte predominante de la miel procede del origen u orígenes florales o vegetales designados y si la miel reúne las características del tipo de miel en cuestión. Podrá designarse igualmente según el color y con el nombre de la región geográfica o topográfica si ha sido producida exclusivamente en la región a que se refiere la denominación.
5.2 Contenido neto.
El contenido neto deberá indicarse en peso en el sistema métrico decimal. Podrá expresarse además en otros sistemas.
5.3 Marca registrada o nombre o razón social y domicilio.
Estos datos corresponderán al fabricante, envasador, distribuidor, importador y exportador, según los casos.
5.4 País de origen.
En el caso de miel de importación deberá indicarse el país de origen.
5.5 Número de registro Sanitario de identificación.
6. METODOS DE ANALISIS Y TOMA DE MUESTRAS
Los métodos de análisis y toma de muestras que se describen a continuación se utilizarán incluso como métodos internacionales de arbitraje.
6.1 Determinación del contenido de azúcar reductor.
6.1.1 Principio del método.
El método es una modificación del método Lane y Eynon (1923), que consiste en reducir la modificación de Soxhlet de la solución de Fehling titulándola en punto de ebullición, con una solución de los azúcares reductores de la miel, utilizando azul de metileno como indicador interno. Para lograr la máxima exactitud en este tipo de determinación es preciso que durante el proceso de normalización y en la determinación de los azúcares reductores de la miel, la reducción de la solución de Fehling se realice a volumen constante. Por tanto, es esencial proceder a una titulación preliminar para determinar el volumen de agua, que debe añadirse antes de realizar las determinaciones para cumplir con este requisito.
6.1.2 Reactivos.
6.1.2.1 Modificación de Soxhlet de la solución de Fehling.
Solución A: Disolver 69,28 g. de sulfato cúprico pentahidratado (CuSO4, 5H2O: Pm = 249,71), en agua destilada hasta obtener un litro de solución.
Conservar durante un día antes de proceder a la titulación.
Solución B: Disolver 346 g. de tartrato sódico potásico (C4H4 KNaO6, 4K2O pm = 282,23) y 100 g. de hidróxido de sodio (NaOH) en agua destilada hasta obtener un litro. Filtrar con un filtro preparado de absbesto.
6.1.2.2 Solución patrón de azúcar invertido (acuosa 10 gramos/litro).
Pesar exactamente 9,5 g. de sacarosa pura, añadir 5 ml. de ácido clorhídrico (ClH puro al 36,5 por 100 p/p aproximadamente) y disolver en agua hasta obtener unos 100 ml.; conservar esta solución acidificada durante varios días a temperatura ambiente (siete días aproximadamente entre 12° y 15° C, o tres días entre 20° y 25° C) y diluir después hasta obtener un litro. (N. B.: El azúcar invertido acidificado al 1 por 100 permanece estable durante varios meses.) Neutralizar un volumen apropiado de esta solución con hidróxido de sodio N (40 g/l.) inmediatamente antes de utilizarla y diluir hasta obtener la concentración necesaria (2 g/l.) para la normalización.
6.1.2.3 Solución de azul de metileno.
Disolver 2 g. en agua destilada hasta obtener un litro.
6.1.2.4 Crema de alúmina.
Preparar una solución fría saturada de alumbre (K2SO4AL2 [SO4]3. 24H2O) en agua.
Añadir hidróxido amónico removiendo constantemente hasta obtener una solución alcalina que reaccione con tornasol, dejar que el precipitado sedimente y lavar por decantación con agua hasta que el agua procedente de los lavados, tratada con solución de cloruro de bario, muestre sólo ligeros indicios de sulfato. Verter el agua sobrante y conservar la crema restante en una botella cerrada.
6.1.3 Toma de muestras.
6.1.3.1 Miel líquida o colada.
Si la muestra está libre de gránulos, mezclar perfectamente removiendo o agitando; si se tienen gránulos, meter el envase cerrado en un baño María, sin sumergirlo y calentar durante treinta minutos a 60° C; luego, si es necesario, hasta que la miel se licúe. Es esencial agitar de vez en cuando. Tan pronto como la muestra se licúe, mezclar perfectamente y enfriar rápidamente. Cuando lo que se desea determinar es el bidroximetilfurfural o la diastasa, no se debe calentar la miel. Si hay alguna sustancia extraña como cera, palillos, abejas, partículas de panal, etc., calentar la muestra al baño María hasta 40° C y filtrarla a través de una estopilla, colocada en un embudo con circulación de agua caliente, antes de tomar la muestra.
6.1.3.2 Miel en panales.
Cortar la parte superior del panal, si está operculado y separar completamente la miel del panal filtrándola por un tamiz cuya malla tenga un reticulado cuadrado de 0,500 mm. por 0,500 mm. (Ref. Recomendación de la ISO R 565). Si algunas porciones de panal o cera pasan a través del tamiz, calentar la muestra como se indica en 6.1.3.1. y filtrar a través de una estopilla. Si la miel en el panal está granulada, calentar hasta que la cera se licúe, remover, enfriar y separar la cera.
6.1.4 Procedimiento.
6.1.4 Preparación de la muestra de ensayo. Primer procedimiento (aplicable a mieles que pueden contener sedimentos).
a) Tomar una muestra de 25 g. pesados exactamente, de miel homogeneizada y colocarla en un matraz volumétrico de 100 ml.; añadir cinco ml. de crema de alúmina (6.1.2.4), diluir en agua a 20° C hasta volumen y filtrar.
b) Diluir 10 ml de esta solución en agua destilada hasta obtener 500 ml. (solución diluida de miel).
O bien:
6.1.4.2 Preparación de la muestra de ensayo. Segundo procedimiento.
a) Pesar cuidadosamente una cantidad representativa de unos dos gramos de la muestra de miel homogeneizada, disolver en agua destilada y diluir en un matraz graduado hasta obtener 200 ml. de solución (solución diluida de miel).
6.1.4.3 Normalización de la solución de Fehling modificada.
Normalizar la solución A modificada de Fehling de forma que cinco ml. exactamente (medir con pipeta) mezclados con cinco ml. aproximadamente de la solución B de Fehling, reaccionen completamente con 0,050 g. de azúcar invertido, añadido en forma de 25 ml. de solución diluida de azúcar invertido (2 g/l.).
6.1.4.4 Titulación preliminar.
Al final de la titulación de reducción, el volumen total de los reactivos añadidos deberá ser de 35 ml. Esto se consigue añadiendo el volumen adecuado de agua antes de comenzar la titulación. Puesto que en los criterios de composición de la norma para la miel se especifica que ésta debe contener más de un 60 por 100 de azúcares reductores (calculados como azúcar invertido), es necesaria una titulación preliminar para determinar él volumen de agua que será preciso añadir a una muestra dada para asegurar que la reducción se realice a volumen constante. Para calcular el volumen de agua, que es preciso añadir, se resta de 25 ml. el volumen de solución diluida de miel, que se ha consumido en la titulación preliminar (x ml.).
Verter con una pipeta cinco ml. de solución A de Fehling en un matraz Erlenmeyer de 250 ml., añadir aproximadamente cinco ml. de solución B de Fehling. Añadir siete ml. de agua destilada, un poco de pómez en polvo, u otro regulador adecuado de la ebullición y echar con una bureta unos 15 ml. de solución diluida de miel. Calentar la mezcla fría sobre una tela metálica hasta ebullición y mantener en ebullición moderada durante dos minutos. Añadir un ml. de solución acuosa de azul de metileno al 0.2 por 100, sin interrumpir la ebullición y completar la titulación sin que el tiempo total de ebullición pase de tres minutos, con pequeñas adiciones repetidas de solución diluida de miel, hasta que el indicador pierda el color. Lo que hay que observar es el color del líquido que permanece en la parte superior. Tomar nota del volumen total de solución diluida de miel (x ml.) que se ha utilizado.
6.1.4.5 Determinación.
Calcular la cantidad de agua que es necesario añadir para que, al final de la titulación, el volumen total de los reactivos sea de 35 ml.; para ello restar de 25 ml. la titulación preliminar (x ml.).
Verter con un pipeta cinco ml. de solución A de Fehling en un matraz Erlenmeyer de 250 ml. y añadir aproximadamente cinco ml. de solución B de Fehling.
Añadir (25 — x) ml. de agua destilada, un poco de pómez en polvo u otro regulador adecuado de la ebullición y, de una bureta, todo el volumen menos 1,5 ml. de solución diluida de miel determinada en la titulación preliminar. Calentar la mezcla fría sobre una tela metálica hasta ebullición y mantener en ebullición moderada durante dos minutos. Añadir 1,0 ml. de solución de azul de metileno al 0,2 por 100 sin interrumpir la ebullición y completar la titulación, sin que el tiempo total de ebullición pase de tres minutos, con pequeñas adiciones repetidas de solución diluida de miel hasta que el indicador pierda el color. Tomar nota del volumen total de solución diluida de miel hasta que el indicador pierda el color. Tomar nota del volumen total de solución diluida de miel (y ml.). La diferencia entre titulaciones duplicadas no deberá ser superior a 0,1 ml.
6.1.5 Cálculo y expresión de los resultados.

donde:
C = g. de azúcar invertido por 100 g. de miel (por 100).
p = peso de la muestra de miel utilizada.
y = volumen de solución diluida de mil consumida durante la determinación (ml.).
6.1.6 Notas sobre el procedimiento.
Para la exactitud y reproductibilidad de la determinación es esencial establecer para cada muestra individual cuál es el volumen de agua necesario para obtener un volumen total de mezcla reactiva de 35 ml. El cuadro que se indica a continuación presenta algunos volúmenes típicos que es posible encontrar en la titulación preliminar para los contenidos de incremento de azúcar invertido indicados, en el supuesto de que la muestra de ensayo (6.1.4.1) pese unos 15 g. o que la muestra de ensayo (6.1.4.2) pese unos dos g.
|
Contenido de azúcar invertido – Porcentaje |
Volumen de agua destilada que ha de añadirse – ml. |
|---|---|
| 60 | 8,3 |
| 65 | 9,6 |
| 70 | 10,7 |
| 75 | 11,0 |
6.2 Determinación del contenido aparente de sacarosa,
6.2.1 Principio del método.
Se basa en el método de inversión de Walker (1917).
6.2.2 Reactivos.
6.2.2.1 Modificación de Soxhlet de la solución de Fehling (6.1.2.1).
6.2.2.2 Solución patrón de azúcar invertido (6.1.2.2).
6.2.2.3 Acido clorhídrico (6,34 N acuosa).
6.2.2.4 Solución de hidróxido de sodio (acuosa 5 N).
6.2.2.5 Solución de azul de metileno 2 g/l. (6.1.2.3).
6.2.3 Toma de muestras.
La miel se prepara para la toma de muestras como en 6.1,3.
6.2.4 Procedimiento.
6.2.4.1 Preparación de la muestra de ensayo.
Preparar la muestra de miel como en 6.1.4.1 (a). Diluir 10 ml. de esta solución en agua destilada hasta obtener 250 ml. de solución de miel (para la determinación de la sacarosa) o bien preparar la solución de miel como en 6.1.4.2 (a).
6.2.4.2 Hidrólisis de la muestra de ensayo.
Poner la solución de miel (50 ml.) en un matraz graduado de 100 ml., junto con 25 ml. de agua destilada; calentar la muestra de ensayo hasta una temperatura de 64 °C en un baño María en ebullición. Retirar a continuación el matraz del baño María y añadir 10 ml. de ácido clorhídrico 6,34 N. Dejar que la solución se enfríe de un modo natural durante quince minutos, y a continuación calentarla hasta 20 ºC y neutralizarla con hidróxido de sodio 5 N, empleando tornasol como indicador. Enfriar de nuevo y completar el volumen hasta 100 ml. (solución diluida de miel).
6.2.4.3 Titulación.
Como en 6.1.4.4 y 6.1.4.5.
6.2.5. Cálculo y expresión de los resultados.
Calcular el tanto por ciento de azúcar invertido (gramo de azúcar invertido por 100 gramos de miel), después de la inversión, utilizando la misma fórmula que para obtener el tanto por ciento de azúcar invertido antes de la inversión en 6.1.5.
Contenido de sacarosa aparente: Contenido de azúcar invertido después de la inversión menos contenido de azúcar invertido antes de la inversión por 0,95.
El resultado se expresa en gramos de sacarosa aparente/100 gramos de miel.
6.3. Determinación del contenido de humedad.
6.3.1. Principio del método.
Se basa en el método refractométrico de Chataway (1932) revisado por Wedmore (1953).
6.3.2. Aparatos.
Refractómetro.
6.3.3. Toma de muestras.
La miel se prepara para la toma de muestras como en 6.1.3.
6.3.4. Procedimiento.
6.3.4.1. Determinación del índice de refracción.
Determinar el índice de refracción de la muestra de ensayo utilizando un refractómetro a temperatura constante, próxima a los 20 °C. Convertir la lectura en contenido de humedad (por ciento m/m.) utilizando la tabla que se indica a continuación. Si la determinación se hace a una temperatura que no sea 20 °C, convertir la lectura en temperatura patrón de 20 °C, utilizando las correcciones de temperatura que se indican más abajo. En el informe sobre el ensayo deberá especificarse el método empleado.
TABLA PARA LA DETERMINACION DEL CONTENIDO DE HUMEDAD
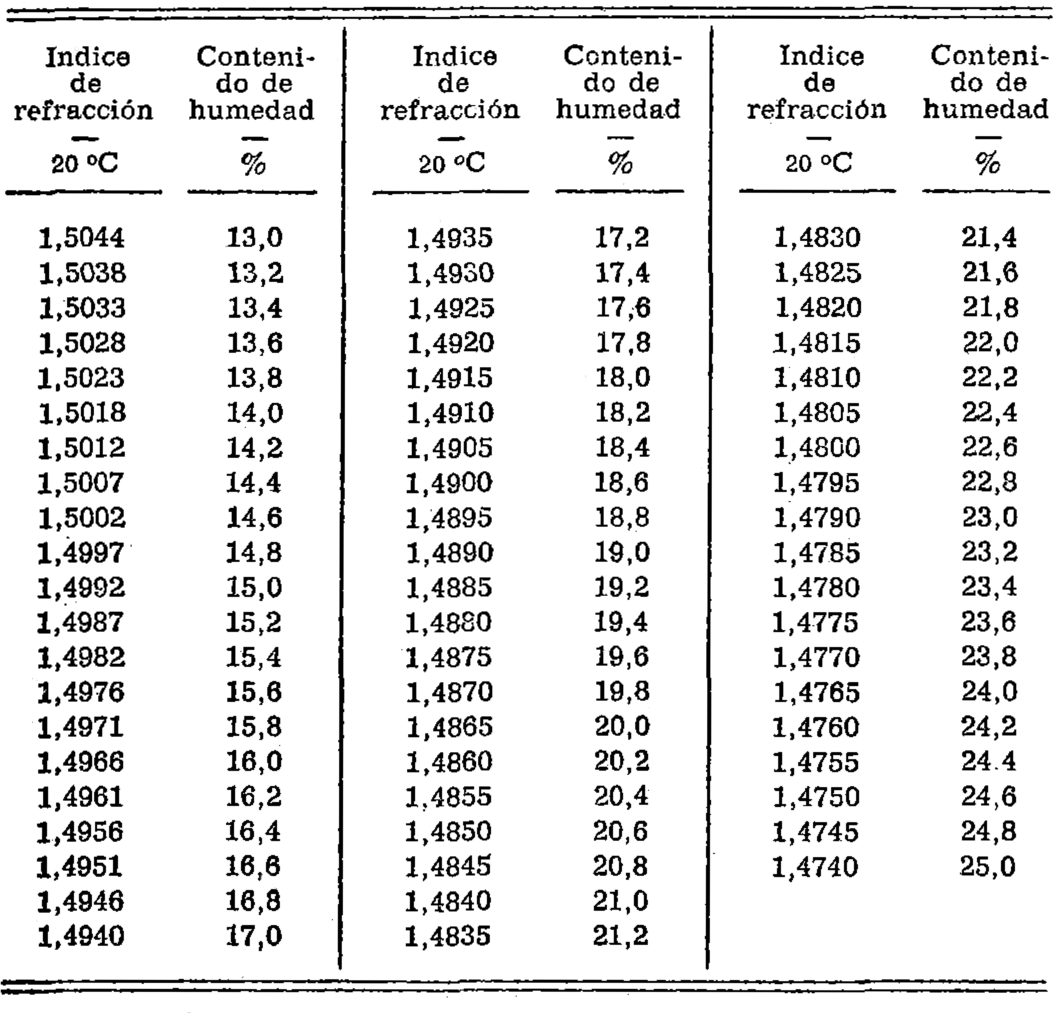
6.3.4.2 Correcciones de temperatura: Indice de refracción.
Temperaturas superiores a 20 °C: Añadir 0,00023 por °C.
Temperaturas inferiores a 20 °C: Restar 0,00023 por ºC.
6.4 Determinación gravimétrica del contenido de sólidos insolubles en agua.
6.4.1 Toma de muestras.
La miel se prepara para la toma de muestras como en 6.1.3.
6.4.2 Procedimiento.
6.4.2.1 Preparación de la muestra de ensayo.
Pesar la miel (20 g.) con precisión al centigramo más próximo (10 mg.) y disolverla en una cantidad adecuada de agua destinada a 80° C y mezclar bien.
6.4.2.2 Determinación gravimétrica.
Filtrar la muestra de ensayo a través de un crisol fino de vidrio sintetizado (tamaño de los poros 15-40 micras) previamente secado y tarado y lavarlo a fondo con agua caliente (80° C) hasta eliminar los azúcares (ensayo de Mohr). Dejar secar el crisol durante una hora a 135° C, enfriar y pesar con una aproximación de 0,1 mg.
6.4.3 Expresión de los resultados.
Los resultados se expresan en tanto por ciento de sólidos insolubles en agua (m/m.).
6.5 Determinación del contenido en sustancias minerales (cenizas).
6.5.1 Toma de muestras.
La miel se prepara para la toma de muestras como en 6.1.3.
6.5.2 Procedimiento.
6.5.2.1 Calcinación de la miel.
La miel (5-10 g.) se pesa exactamente y se coloca en una cápsula de platino o de sílice calcinada, previamente pesada, y se calienta suavemente en un horno de mufla hasta que la muestra se ennegrezca y seque y no haya peligro de pérdidas por formación dé espuma y rebosamiento. Puede utilizarse también una lámpara de rayos infrarrojos para carbonizar la muestra antes de introducirla en el homo. En caso necesario, pueden añadirse unas gotas de aceite de oliva, para impedir la formación de espuma. A continuación, calcinar la muestra a 600° C, hasta peso constante. La muestra se enfría antes de pesarla.
6.5.3 Expresión de los resultados.
Los resultados se expresan en tanto por ciento de cenizas (m/m.).
6.6 Determinación de la acidez.
6.6.1 Toma de muestras.
La miel se prepara para la toma de muestras como en 6.1.3.
6.6.2 Reactivos.
6.6.2.1 Hidróxido de sodio 0,1 N (libre de carbonatos).
6.6.2.2 Indicador de fenolftaleína al 1 por 100 (m/.) en etanol neutralizado.
6.6.2.3 Agua destilada, previa extracción del dióxido de carbono, hirviéndola y enfriándola a continuación.
6.6.3 Procedimiento.
6.6.3.1 Preparación de la muestra de ensayo.
Pesar la miel (10,0 g.) y disolverla en 75 ml. de agua destilada (6.6.2.3).
6.6.3.2 Titulación.
Titular la muestra de ensayo con solución de hidróxido de sodio 0,1 N libre de carbonatos, utilizando como indicador 4 ó 5 gotas de fenolftaleína. El color del punto final deberá persistir durante diez segundos. Para las muestras de color oscuro se tomará menor cantidad. Otro modo de proceder consistirá en utilizar un pH-metro y titular la muestra hasta pH 8,3.
6.6.4 Cálculo y expresión de los resultados.
Los resultados se expresan en milivales (miliequivalentes) de ácido/kg. de miel, y se calculan en la forma siguiente:
Acidez = 10 v.
donde v = el número de ml. de NaOH 0,1 N utilizados en la neutralización de 10 g. de miel.
6.7 Determinación de la actividad de la diastasa.
6.7.1 Principio del método
Se basa en el método de Schade y otros (1958), modificado por White y otros (1959) y por Hadorn (1961).
6.7.2 Reactivos.
6.7.2.1 Solución madre de yodo.
Disolver 8,8 g. de yodo de calidad para análisis en 30-40 ml. de agua que contenga 22 g. de yoduro de potasio de calidad para análisis, y diluir con agua hasta obtener un litro.
6.7.2.2 Solución de yodo 0,0007 N.
Disolver 20 g. de yoduro de potasio de calidad para análisis en 30-40 ml. de agua en un matraz volumétrico de 500 ml. Añadir 5,0 ml. de solución madre de yodo y completar hasta volumen. Preparar una solución nueva en días alternos.
6.7.2.3 Amortiguador de acetato-pH 5,3 (1,59 M).
Disolver 87 g. de acetato de sodio 3H20 en 400 ml. de agua, añadir unos 10,5 ml. de ácido acético glacial disuelto en un poco de agua y completar hasta 500 ml. Ajustar el pH a 5,3 con acetato de sodio o ácido acético, según el caso, utilizando un pH-metro.
6.7.2.4 Solución de cloruro de sodio 0,5 M.
Disolver 14,5 g. de cloruro de sodio de calidad para análisis en agua destilada hervida y completar hasta 500 ml. El tiempo de conservación está limitado por la formación de mohos.
6.7.2.5 Solución de almidón.
Emplear un almidón con un índice de azul comprendido entre 0,5-0,55 utilizando una célula de 1 cm.; para determinar el índice de azul utilícese el método descrito más abajo.
Pesar una cantidad de almidón equivalente a 2,0 g. de almidón anhidro. Mezclar con 90 ml. de agua en un matraz cónico de 250 ml. Ponerla a hervir rápidamente, agitando la solución todo lo posible, calentando sobre una malla de alambre, preferiblemente con el centro de asbesto. Hervir suavemente durante tres minutos, tapar y dejar enfriar, espontáneamente hasta la temperatura ambiente. Transvasar a un matraz volumétrico de 100 ml., poner el matraz en un baño María a 40 °C hasta que el líquido alcance esa temperatura y completar hasta volumen, a 40 °C.
Método para determinar el índice de azul de almidón: Disolver por el método anterior una cantidad de almidón equivalente a 1 g. de almidón anhidro, enfriar la solución, añadir, 2,5 ml. de amortiguador de acetato y completar el volumen hasta 100 ml. en un matraz volumétrico.
Echar, en un matraz volumétrico de 100 ml. 75 ml. de agua, 1 ml. de ácido clorhídrico N y 1,5 ml. de solución de yodo 0,02 N. A continuación añadir 0,5 ml. de solución de almidón y completar con agua hasta volumen. Dejar reposar una hora en la oscuridad y leer después en un espetrofotómetro a 660 nm. empleando una célula de 1 cm. y un testigo que contenga todos los ingredientes anteriores, excepto la solución de almidón.
Lectura en la escala de absorbancia = índice de azul.
6.7.3 Aparatos,
6.7.3.1 Baño María a 40° ± 0,2 °C.
6.7.3.2 Espectrofotómetro que permita leer a 660 nm.
6.7.4 Toma de muestras.
La muestra de miel se prepara como en 6.1.3, sin calentar.
6.7.5 Procedimiento.
6.7.5.1 Preparación de las muestras de ensayo.
Solución de miel: Poner 10,0 g. (pesados) de miel en un vaso de precipitados de 50 ml. y añadir 5,0 ml. de solución de amortiguador de acetato y 20 ml. de agua para disolver la muestra. Disolver completamente la muestra agitando la solución fría. Echar 3,0 ml. de solución de cloruro de sodio en un matraz aforado de 50 ml., pasar a este matraz la muestra de miel disuelta y completar el volumen hasta 50 ml.
N. B. Es esencial que la miel esté amortiguada antes de entrar en contacto con el cloruro de sodio.
Normalización de la solución de almidón: Calentar la solución de almidón a 40 °C y, mediante una pipeta, echar 5 ml. de esa solución en 10 ml. de agua a 40 °C y mezclar bien. Con una pipeta verter 1 ml. de esta última solución en 10 ml. de solución de yodo 0,0007 N diluida en 35 ml. de agua y mezclar bien. Leer la coloración a 660 nm., contra un testigo de agua, utilizando una célula de 1 cm. La absorbancia debe ser 0,760 ± 0,020. En caso necesario, ajustar el volumen de agua añadido hasta obtener la absorbancia exacta.
6.7.5.2 Determinación de la absorbancia.
Mediante una pipeta, verter 10 ml. de solución de miel en un vaso cilindrico graduado de 50 ml. y colocarlo en un baño María a 40 °C + 0,2 °C, junto con el matraz que contiene la solución de almidón en la solución de miel, mezclar y poner en marcha un cronómetro. A intervalos de cinco minutos sacar porciones de 1 ml. y echarlas en 10,00 ml. de solución de yodo 0,0007 N. Mezclar y diluir hasta volumen normalizado (véase 6.7.5.1). Determinar inmediatamente la absorbancia a 660 nm., en el espectrofotómetro, empleando una célula de 1 cm. Seguir tomando porciones de 1 ml. a intervalos, hasta lograr una absorbancia menor de 0,235.
6.7.6 Cálculo y expresión de los resultados.
Representar gráficamente la absorbancia en función del tiempo (minutos) sobre un papel cuadriculado. Trazar una línea recta que una, por lo menos, los tres últimos puntos del gráfico para determinar el momento en que la mezcla de la reacción alcanza una absorbancia de 0,235. Dividir 300 por el tiempo en minutos para obtener el índice de diastasa (ID). Este número expresa la actividad de la diastasa en ml. de solución de almidón al l por 100 hidrolizada por la enzima contenida en 1 g. de miel, en una hora, a 40 °C. Este índice de diastasa corresponde al número de la Escala Gothe.
Actividad de la diastasa = ID = ml. de solución de almidón (1 por 100)/g. de miel/h. a 40 °C.
6.8 Determinación fotométrica del contenido de hidroximetilfurfural (H. M. F.).
(Este método podrá sustituirse en el futuro por un método espectrofotométrico.)
6.8.1 Principio del método.
Se basa en el método de Winkler (1955).
6.8.2 Reactivos.
6.8.2.1 Solución de ácido barbitúrico.
Tomar una porción de 500 mg. de ácido barbitúrico y ponerla en un matraz graduado de 100 ml., empleando 70 ml. de agua. Colocar en un baño maría caliente hasta que se disuelva, enfriar y completar hasta volumen.
6.8.2.2 Solución de p-toluidina.
Tomar una porción de 10,0 g. de p-toluidina, de calidad para análisis, y disolverla en unos 50 ml. de isopropanol, calentando suavemente en baño maría. Transvasar a un matraz graduado de 100 ml. con isopropanol y añadir 10 ml. de ácido acético glacial. Enfriar y completar hasta volumen con isopropanol. Conservar la solución en un lugar oscuro. No emplearla hasta que por lo menos hayan transcurrido veinticuatro horas.
6.8.2.3 Agua destilada (libre de oxígeno).
Hacer pasar nitrógeno gaseoso a través de agua destilada en ebullición. Después enfriar el agua.
6.8.3 Aparatos.
6.8.3.1 Espectrofotómetro que permita leer a 550 nm.
6.8.3.2 Baño María.
6.8.4 Toma de muestras.
La miel se prepara como en 6.1.3, sin calentar.
6.8.5 Procedimiento.
6.8.5.1 Preparación de la muestra de ensayo.
Pesar 10 g. de la muestra de miel y disolver, sin calentar, en 20 ml. de agua destilada libre de oxígeno (6.8.2.3). Transvasar esta solución a un matraz graduado de 50 ml. y completar hasta volumen (solución de miel). La muestra deberá someterse a ensayo inmediatamente después de preparada.
6.8.5.2 Determinaciones fotométricas.
Con una pipeta, verter 2,0 ml. de solución de miel en cada une de dos tubos de ensayo y añadir en cada uno de ellos 5,0 ml. de solución de p-toluidina. Echar en uno de los tubos, con una pipeta, 1 ml. de agua y, en otro, 1 ml. de solución de ácido barbitúrico y agitar ambas mezclas. El tubo, al que se ha añadido agua, sirve como testigo. La adición de los reactivos debe hacerse ininterrumpidamente y terminarse en uno o dos minutos.
Inmediatamente después de haber alcanzado el valor máximo (entre tres a cuatro minutos, después de la adición del ácido barbitúrico), leer la absorbancia de la muestra contra el testigo a 550 nm., empleando una célula de 1 cm.
6.8.6 Expresión de los resultados.
Los resultados se expresan en mg. de H. M. F. por 100 g. de miel y el contenido de H. M. F. se calcula por la fórmula siguiente:

6.8.7 Comprobación del método.
El método se verificará utilizando una solución patrón de hidroximetilfurfural (H. M. F.) normalizada, disolviendo H. M. F. comercial o preparado en laboratorio y ensayando espectrofotométricamente, donde ε = 16,830 (J. H. Turnes, 1954) a 284 nm., empleando patrones, de 0-300 μ g.
6.9 Determinación de las dextrinas.
Pesar ocho gramos en un matraz de 100 ml., disolver en seis ml. de agua. Enrasar con alcohol, agitando constantemente.
Dejar un tiempo para que precipiten las dextrinas.
Decantar el líquido claro a través de un filtro y lavar el residuo que quede en el matraz con 10 ml. de alcohol, pasándolo a través del mismo filtro.
Disolver la dextrina con agua hirviendo y filtrar a través del filtro ya usado. Recoger este último filtrado (disolución de las dextrinas) en un pesasustancias, lavando el filtro con pequeños volúmenes de agua caliente. Evaporar a sequedad hasta peso constante (procurar no subir demasiado la temperatura)
El precipitado, después de seco, se disuelve en agua y se diluye a un volumen determinado (50 ml./0,5 g. de precipitado).
En la solución se determinan los azúcares reductores, posteriormente se hidroliza y se vuelven a determinar los azúcares.
Agència Estatal Butlletí Oficial de l'Estat
Avda. de Manoteras, 54 - 28050 Madrid




